

Seed amplification and neurodegeneration marker trajectories in individuals at risk of prion disease
- PMID: 36975162
- PMCID: PMC10232278
- DOI: 10.1093/brain/awad101
Seed amplification and neurodegeneration marker trajectories in individuals at risk of prion disease
Abstract
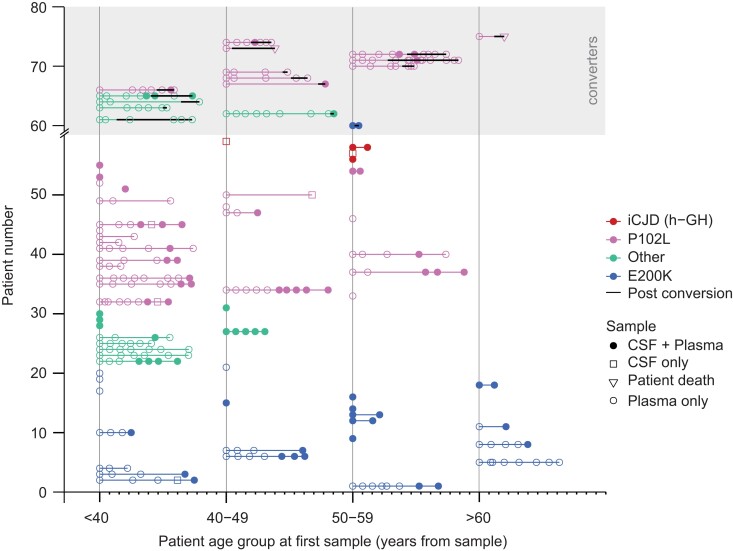
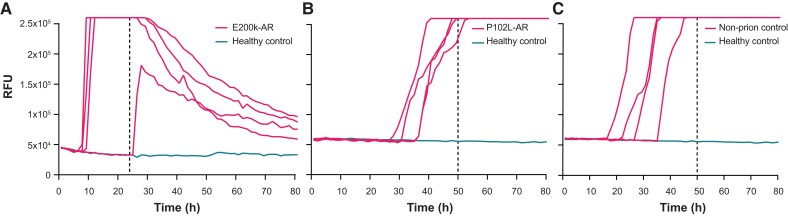
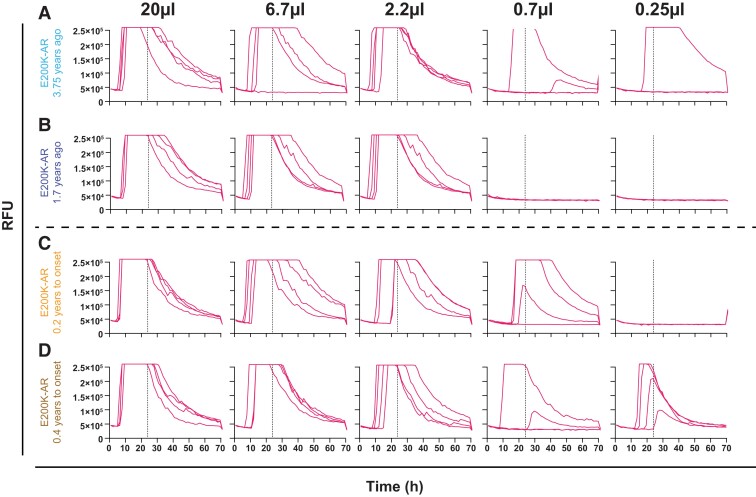
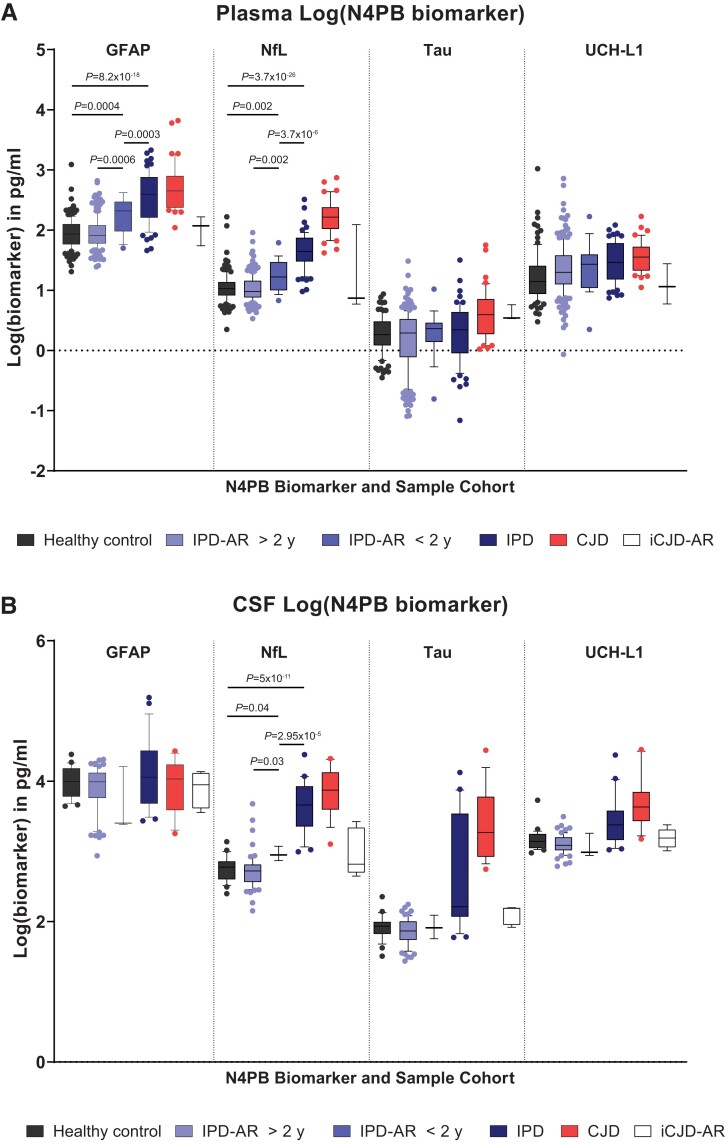
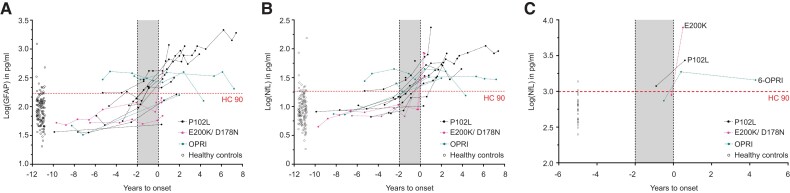
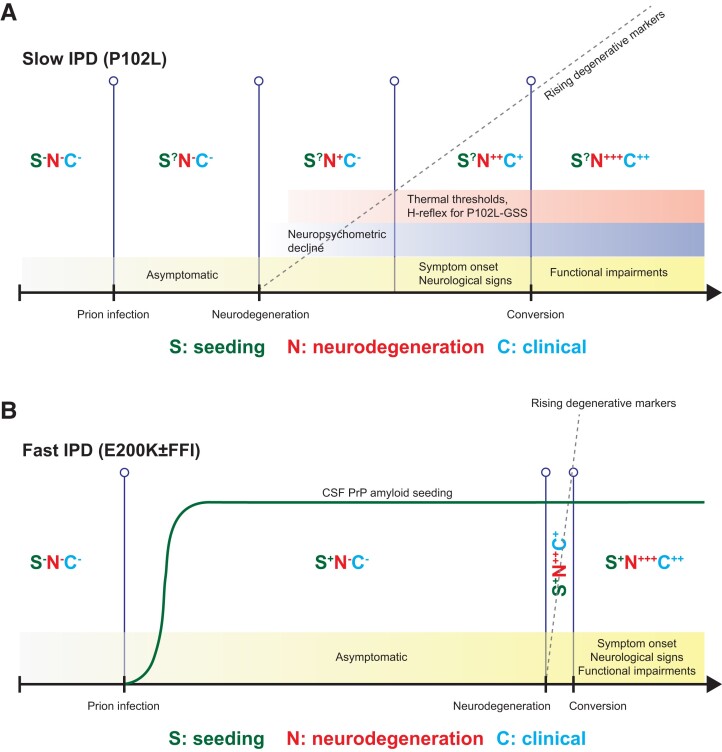
Human prion diseases are remarkable for long incubation times followed typically by rapid clinical decline. Seed amplification assays and neurodegeneration biofluid biomarkers are remarkably useful in the clinical phase, but their potential to predict clinical onset in healthy people remains unclear. This is relevant not only to the design of preventive strategies in those at-risk of prion diseases, but more broadly, because prion-like mechanisms are thought to underpin many neurodegenerative disorders. Here, we report the accrual of a longitudinal biofluid resource in patients, controls and healthy people at risk of prion diseases, to which ultrasensitive techniques such as real-time quaking-induced conversion (RT-QuIC) and single molecule array (Simoa) digital immunoassays were applied for preclinical biomarker discovery. We studied 648 CSF and plasma samples, including 16 people who had samples taken when healthy but later developed inherited prion disease (IPD) ('converters'; range from 9.9 prior to, and 7.4 years after onset). Symptomatic IPD CSF samples were screened by RT-QuIC assay variations, before testing the entire collection of at-risk samples using the most sensitive assay. Glial fibrillary acidic protein (GFAP), neurofilament light (NfL), tau and UCH-L1 levels were measured in plasma and CSF. Second generation (IQ-CSF) RT-QuIC proved 100% sensitive and specific for sporadic Creutzfeldt-Jakob disease (CJD), iatrogenic and familial CJD phenotypes, and subsequently detected seeding activity in four presymptomatic CSF samples from three E200K carriers; one converted in under 2 months while two remain asymptomatic after at least 3 years' follow-up. A bespoke HuPrP P102L RT-QuIC showed partial sensitivity for P102L disease. No compatible RT-QuIC assay was discovered for classical 6-OPRI, A117V and D178N, and these at-risk samples tested negative with bank vole RT-QuIC. Plasma GFAP and NfL, and CSF NfL levels emerged as proximity markers of neurodegeneration in the typically slow IPDs (e.g. P102L), with significant differences in mean values segregating healthy control from IPD carriers (within 2 years to onset) and symptomatic IPD cohorts; plasma GFAP appears to change before NfL, and before clinical conversion. In conclusion, we show distinct biomarker trajectories in fast and slow IPDs. Specifically, we identify several years of presymptomatic seeding positivity in E200K, a new proximity marker (plasma GFAP) and sequential neurodegenerative marker evolution (plasma GFAP followed by NfL) in slow IPDs. We suggest a new preclinical staging system featuring clinical, seeding and neurodegeneration aspects, for validation with larger prion at-risk cohorts, and with potential application to other neurodegenerative proteopathies.
Keywords: GFAP; NfL; RT-QuIC; inherited; prion.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
H.Z. has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Passage Bio, Pinteon Therapeutics, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. J.B.R. has provided consultancy and/or served on advisory boards for Asceneuron, Astex, Astonautx, Curasen, SV Health, UCB, and Wave. J.C. is a director and shareholder of D-Gen, an academic spinout in the field of prion disease diagnosis and therapeutics. The other authors report no competing interests.
Figures
Comment in
-
Where have prions been all our lives?Brain. 2023 Jun 1;146(6):2206-2207. doi: 10.1093/brain/awad143. Brain. 2023. PMID: 37161596 Free PMC article.
References
-
- Collinge J. Prion diseases of humans and animals: Their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. - PubMed
-
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. - PubMed
-
- Mead S, Lloyd S, Collinge J. Genetic factors in mammalian prion diseases. Annu Rev Genet. 2019;53:117–147. - PubMed
-
- Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14:273–281. - PubMed
-
- Collinge J. Variant creutzfeldt-jakob disease. Lancet. 1999;354:317–323. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
