

Disruption of the MYC Superenhancer Complex by Dual Targeting of FLT3 and LSD1 in Acute Myeloid Leukemia
- PMID: 36976323
- PMCID: PMC10330306
- DOI: 10.1158/1541-7786.MCR-22-0745
Disruption of the MYC Superenhancer Complex by Dual Targeting of FLT3 and LSD1 in Acute Myeloid Leukemia
Abstract
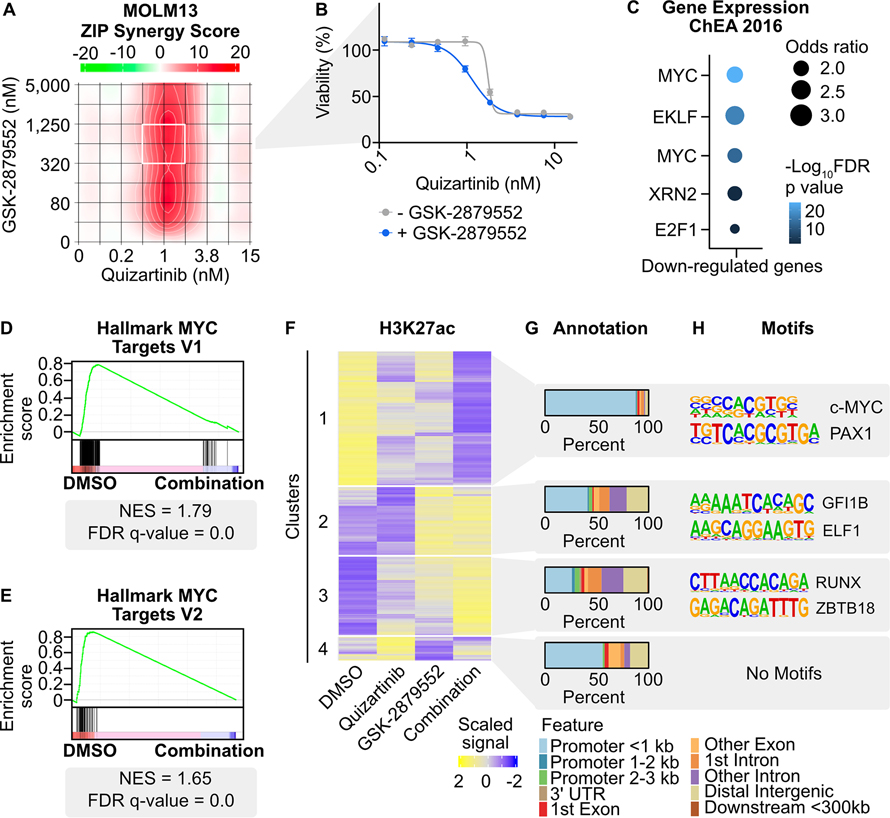
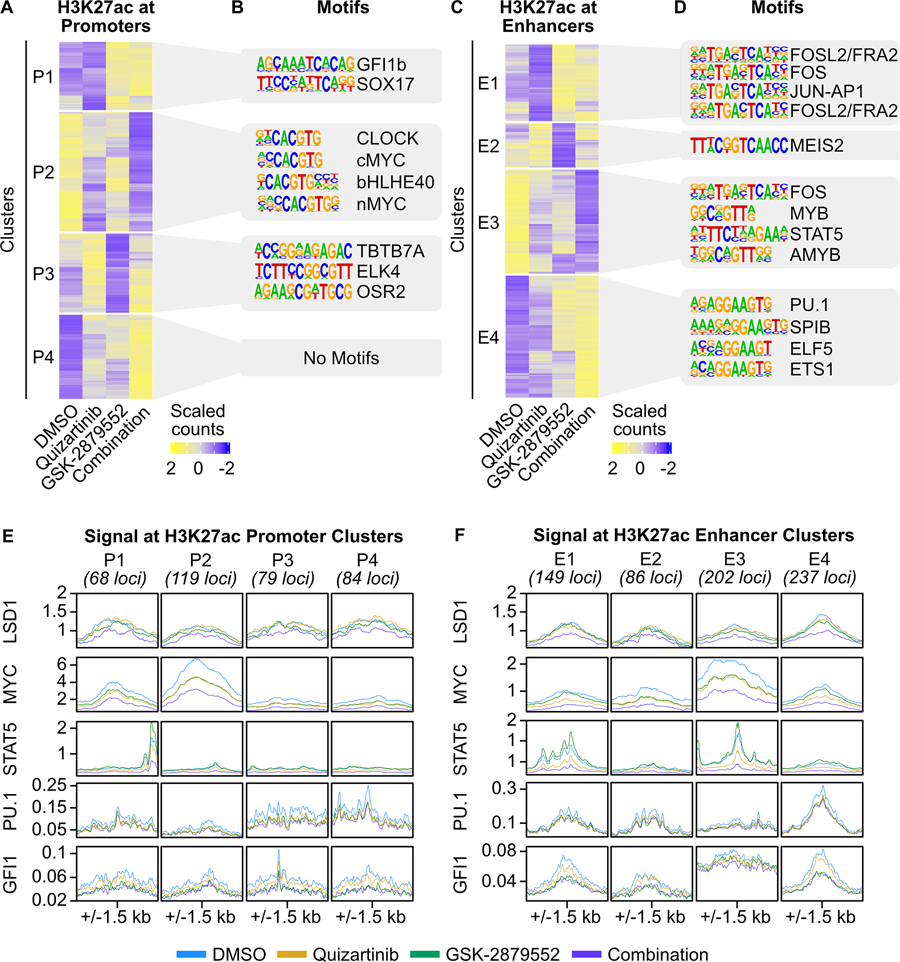
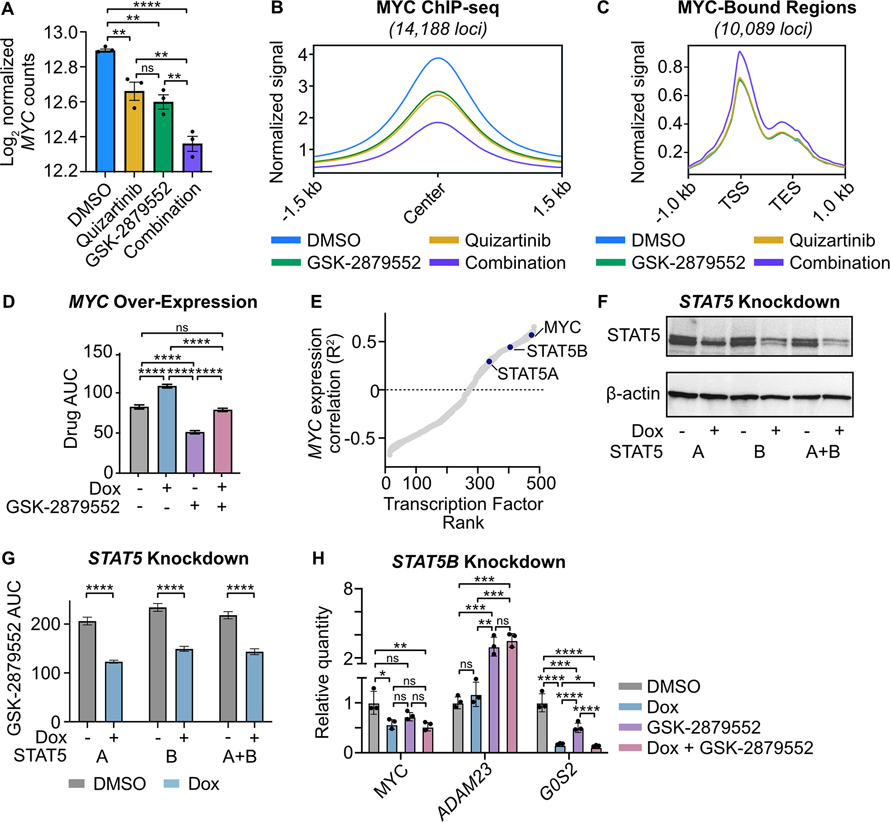
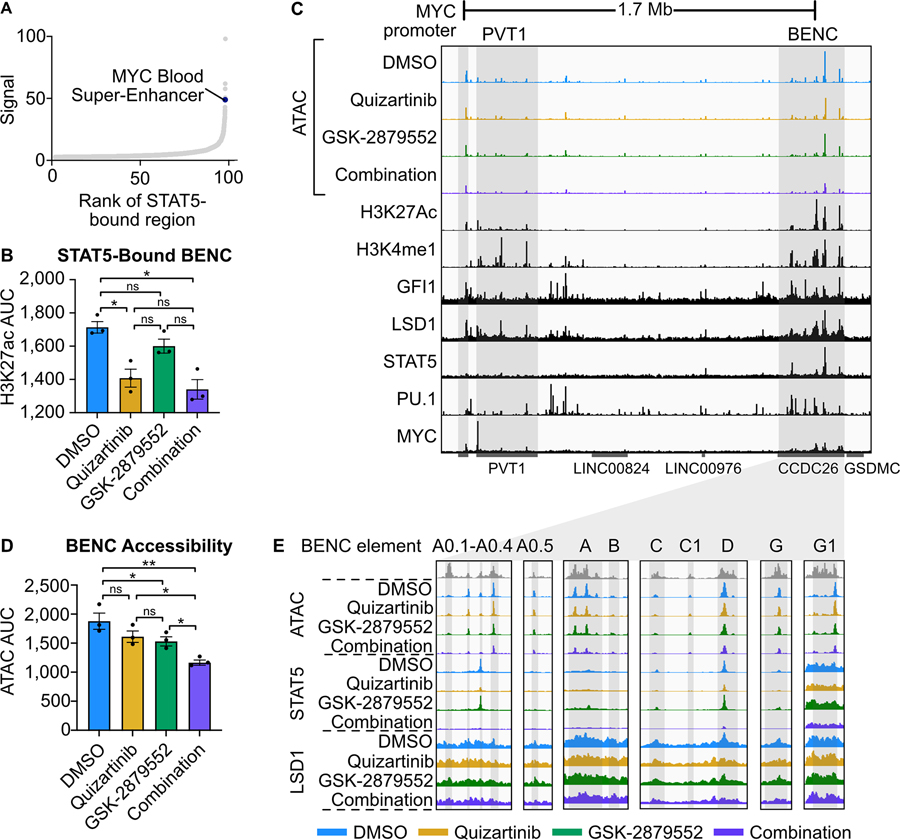
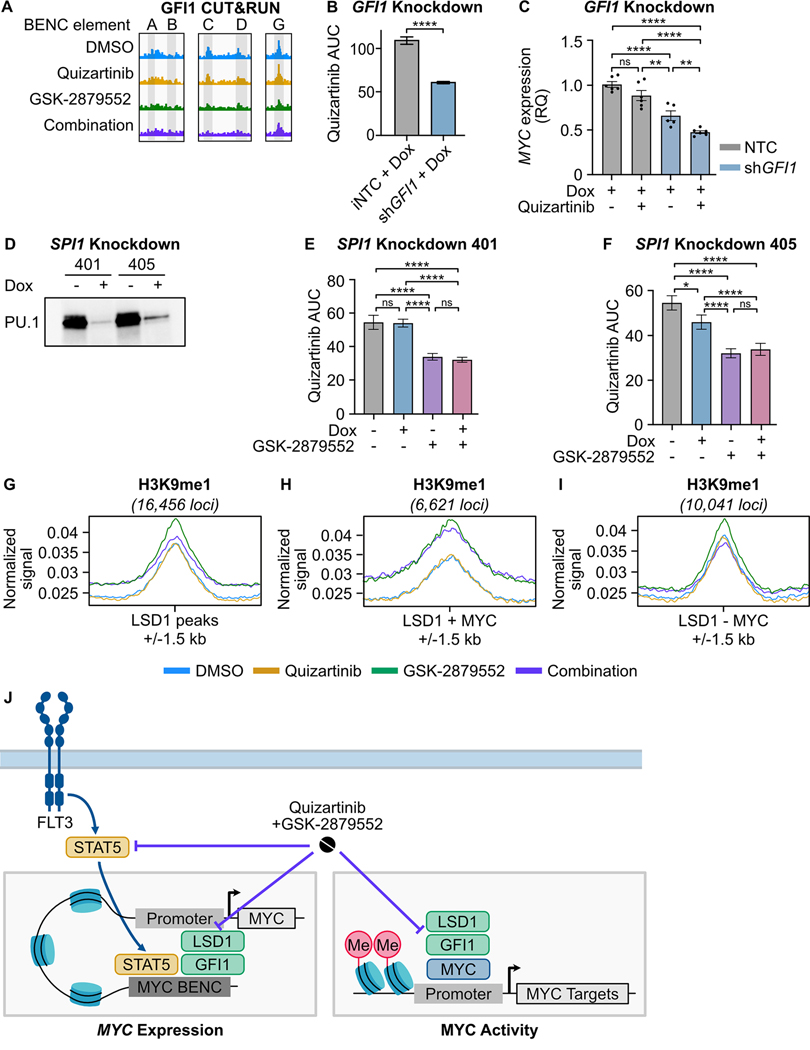
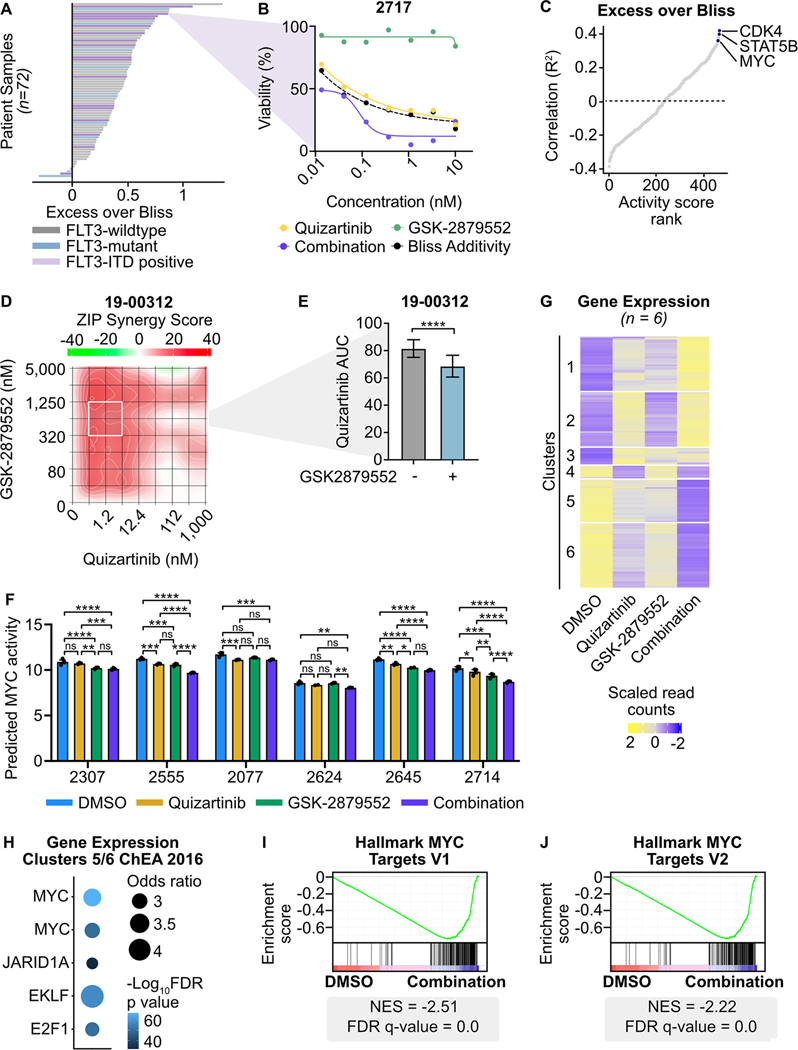
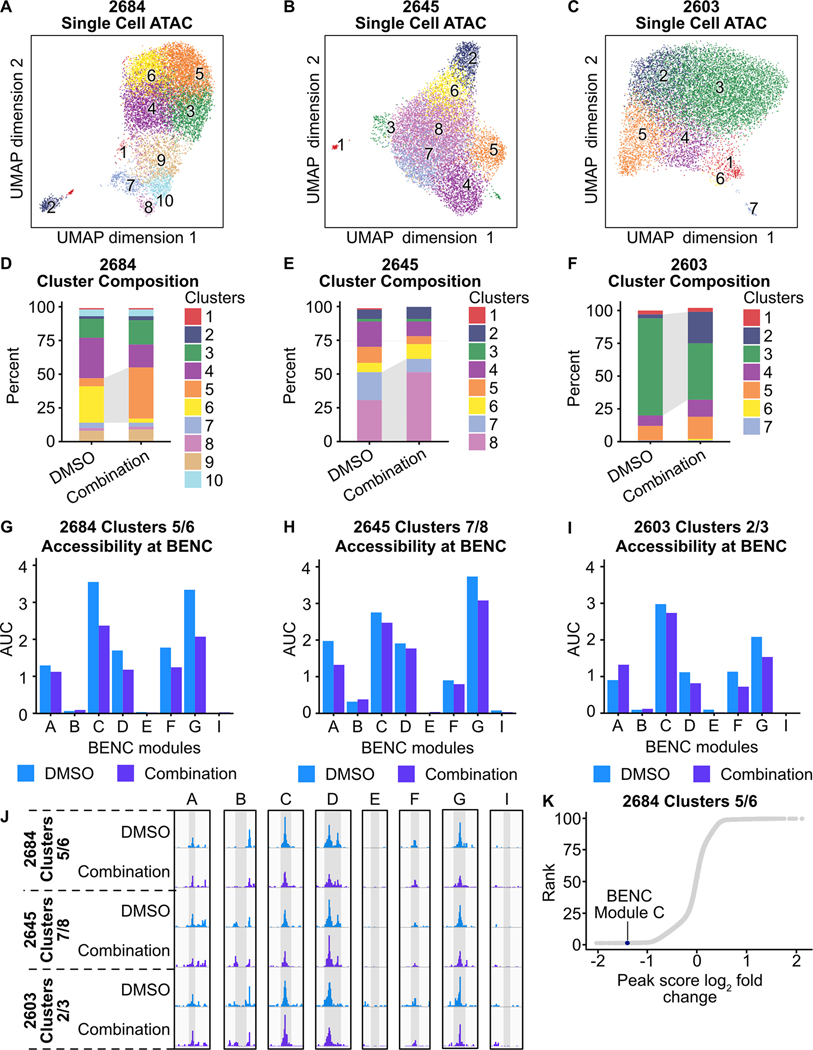
Mutations in Fms-like tyrosine kinase 3 (FLT3) are common drivers in acute myeloid leukemia (AML) yet FLT3 inhibitors only provide modest clinical benefit. Prior work has shown that inhibitors of lysine-specific demethylase 1 (LSD1) enhance kinase inhibitor activity in AML. Here we show that combined LSD1 and FLT3 inhibition induces synergistic cell death in FLT3-mutant AML. Multi-omic profiling revealed that the drug combination disrupts STAT5, LSD1, and GFI1 binding at the MYC blood superenhancer, suppressing superenhancer accessibility as well as MYC expression and activity. The drug combination simultaneously results in the accumulation of repressive H3K9me1 methylation, an LSD1 substrate, at MYC target genes. We validated these findings in 72 primary AML samples with the nearly every sample demonstrating synergistic responses to the drug combination. Collectively, these studies reveal how epigenetic therapies augment the activity of kinase inhibitors in FLT3-ITD (internal tandem duplication) AML.
Implications: This work establishes the synergistic efficacy of combined FLT3 and LSD1 inhibition in FLT3-ITD AML by disrupting STAT5 and GFI1 binding at the MYC blood-specific superenhancer complex.
©2023 American Association for Cancer Research.
Conflict of interest statement
Conflict of Interest
Figures
References
-
- Herold T, Rothenberg-Thurley M, Grunwald VV, Janke H, Goerlich D, Sauerland MC, et al. Validation and refinement of the revised 2017 European LeukemiaNet genetic risk stratification of acute myeloid leukemia. Leukemia [Internet]. Nature Publishing Group; 2020. [cited 2022 Apr 21];34:3161–72. Available from: https://www.nature.com/articles/s41375-020-0806-0 - PMC - PubMed
-
- Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. New England Journal of Medicine. Massachusetts Medical Society; 2019;381:1728–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
