

Loss of Dnmt3a impairs hematopoietic homeostasis and myeloid cell skewing via the PI3Kinase pathway
- PMID: 36976647
- PMCID: PMC10243813
- DOI: 10.1172/jci.insight.163864
Loss of Dnmt3a impairs hematopoietic homeostasis and myeloid cell skewing via the PI3Kinase pathway
Abstract
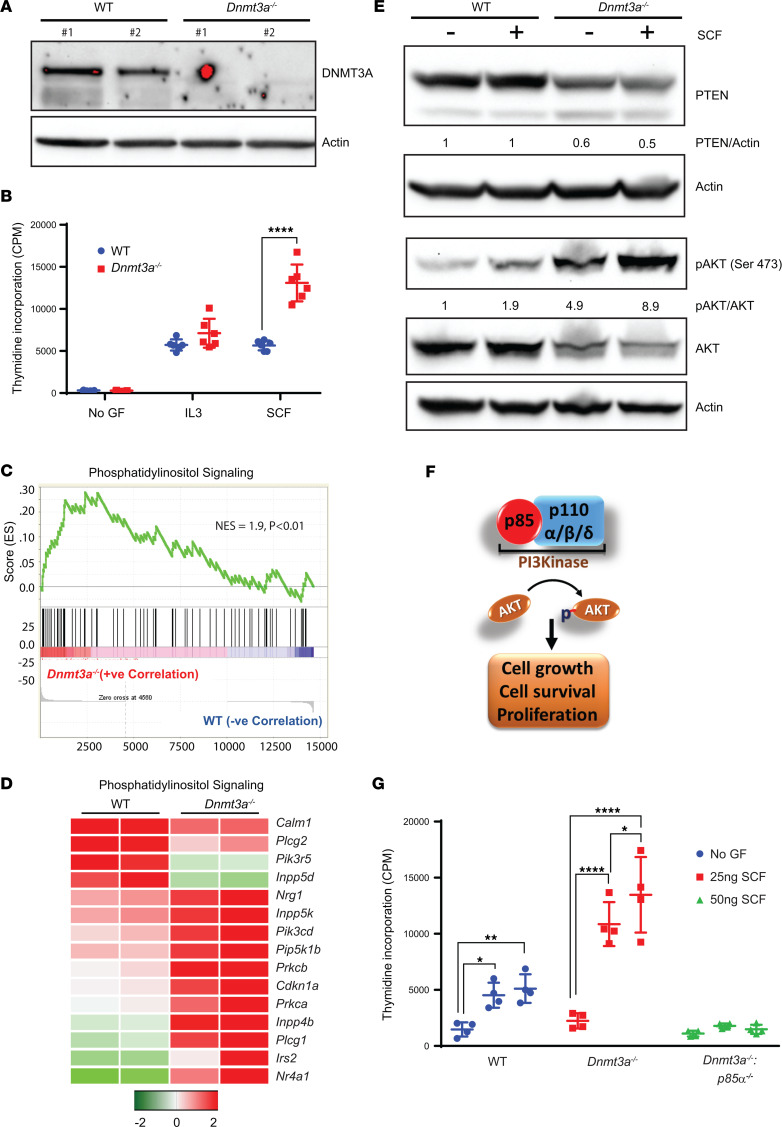
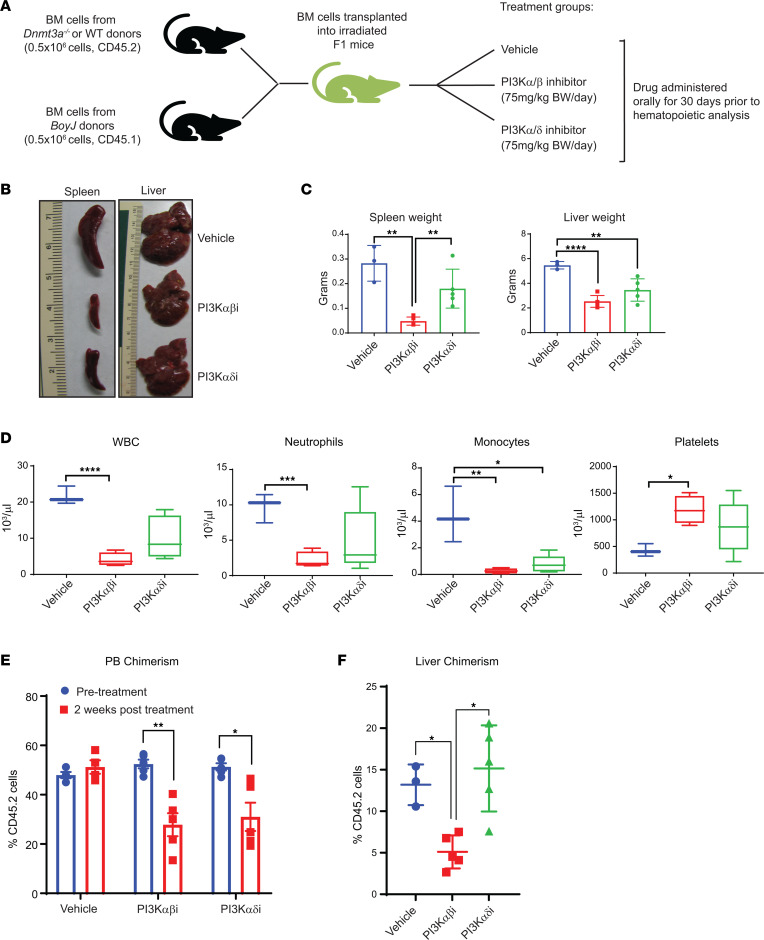
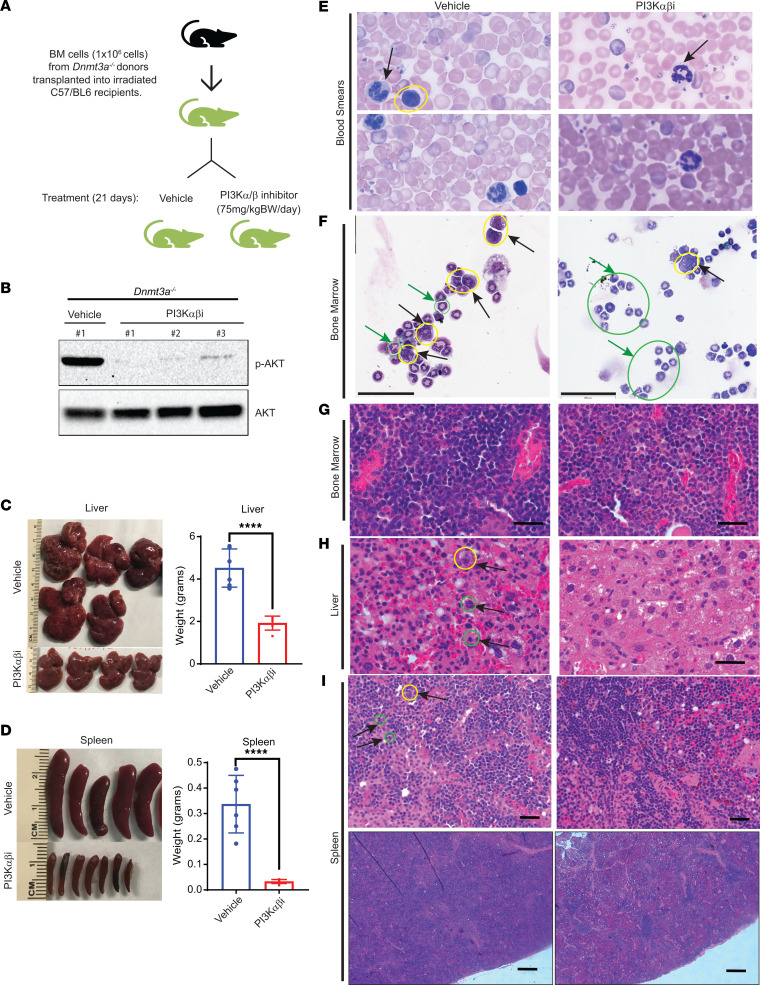
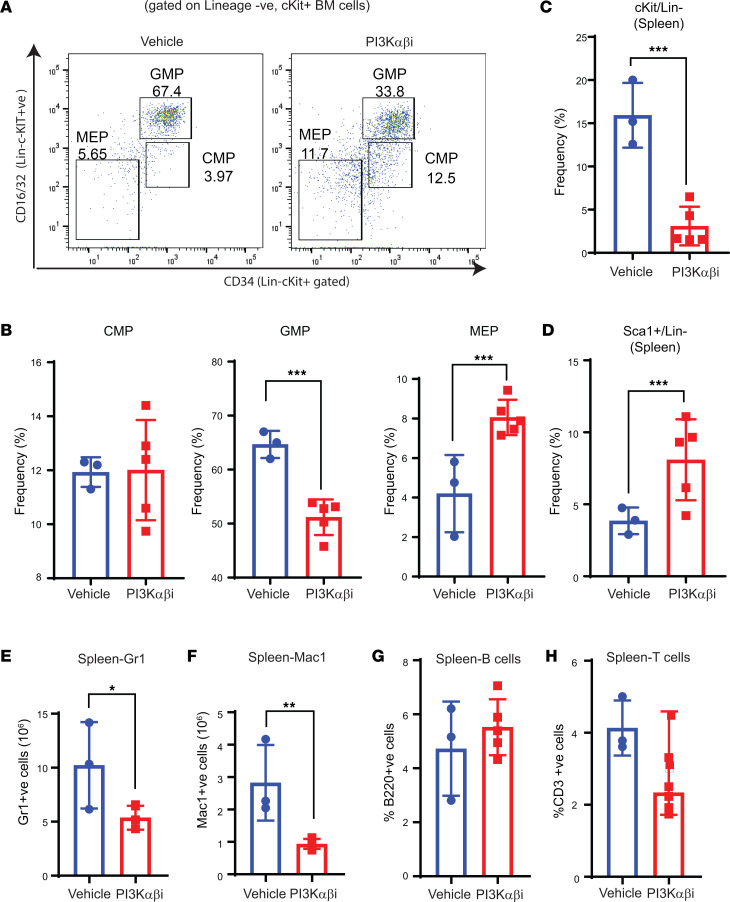
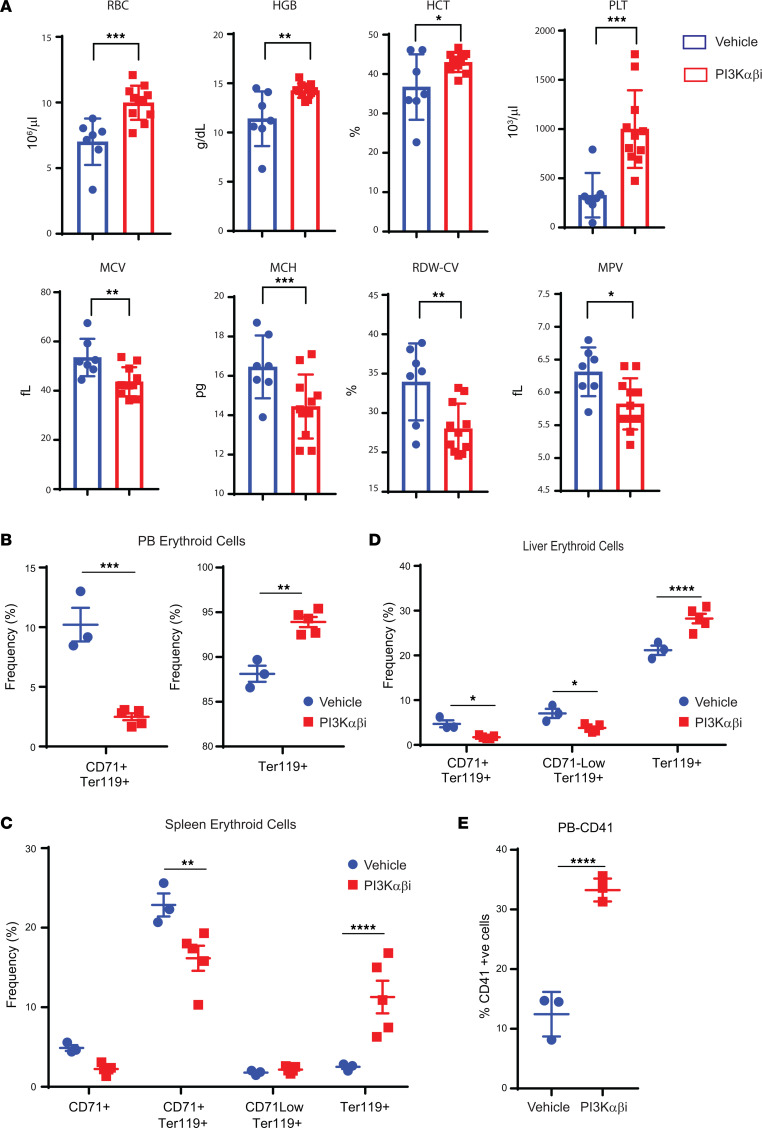
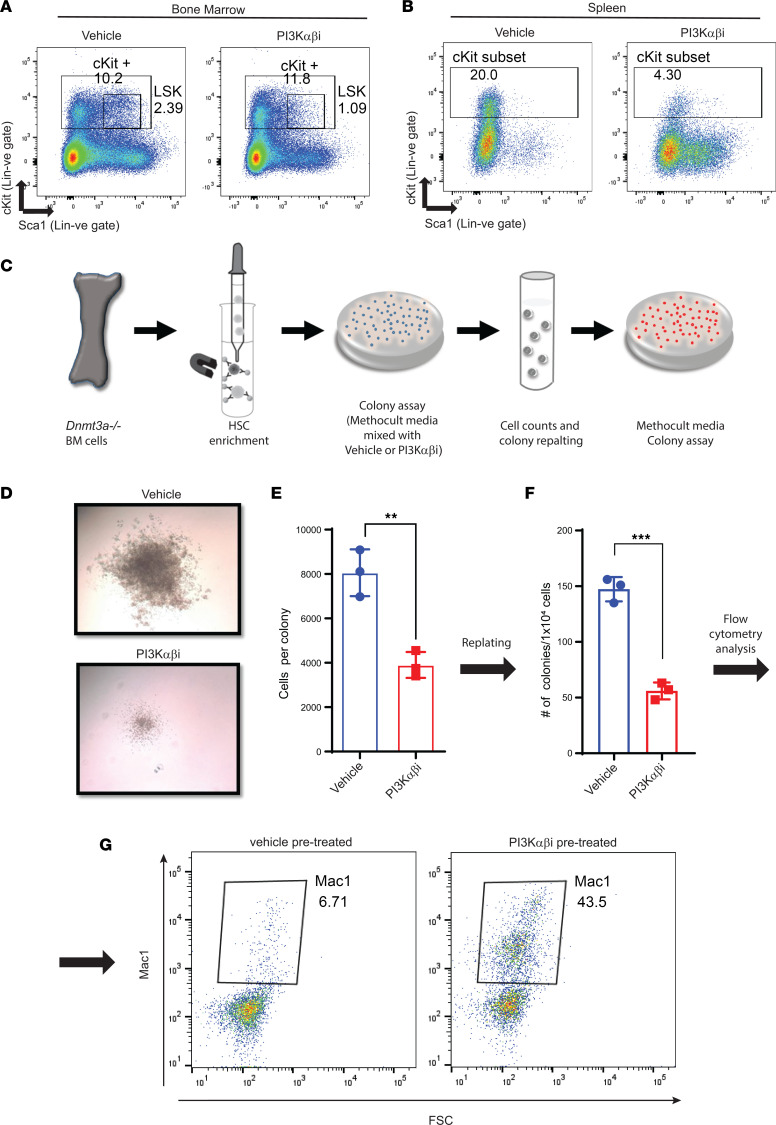
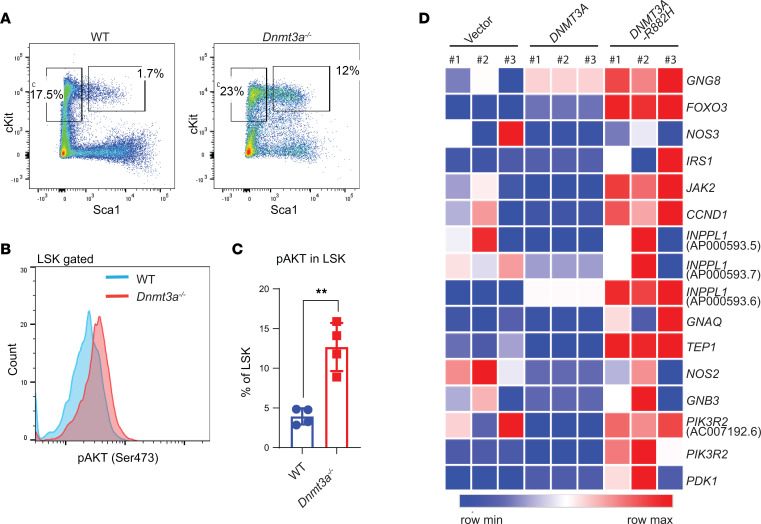
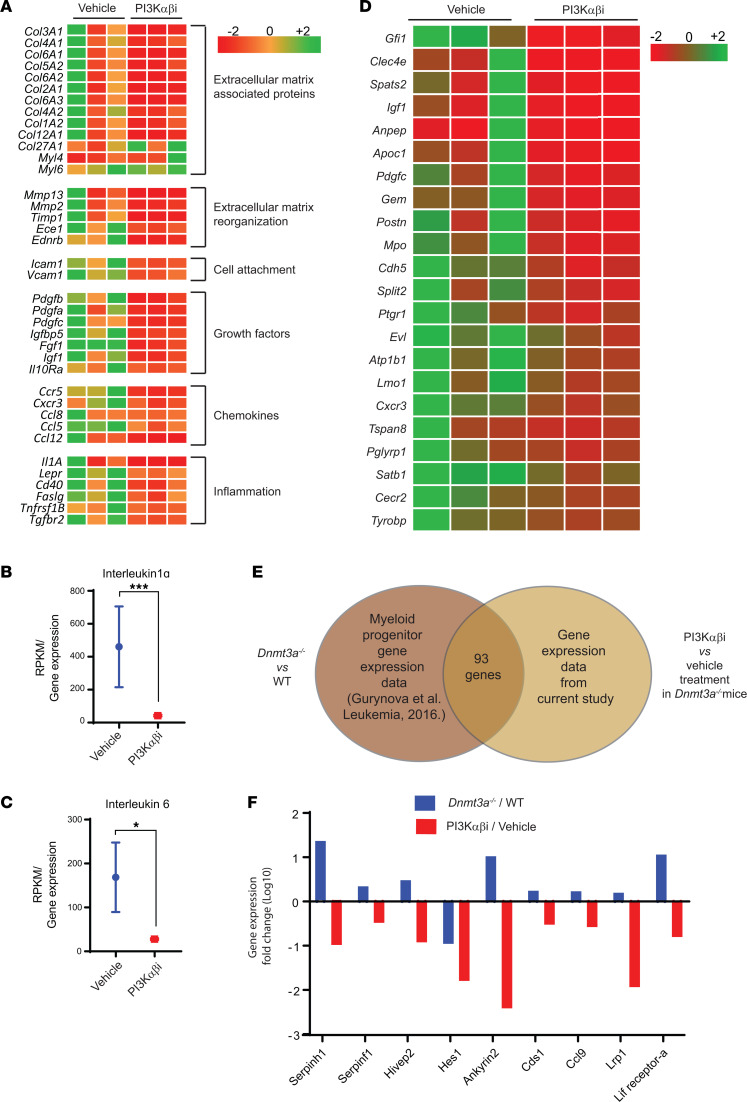
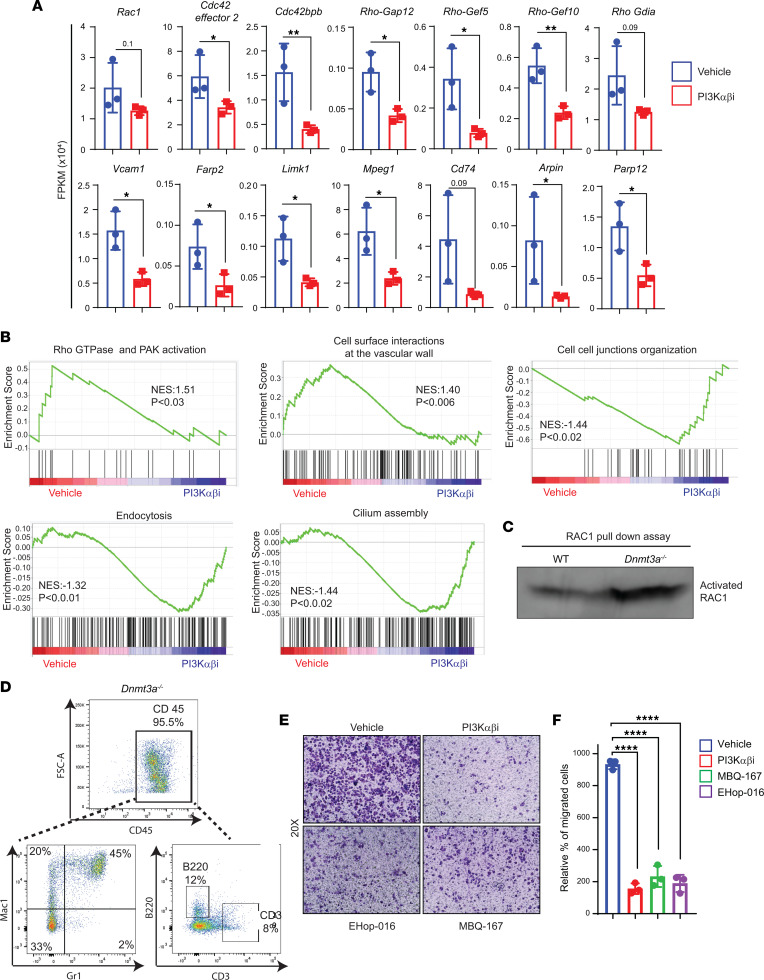
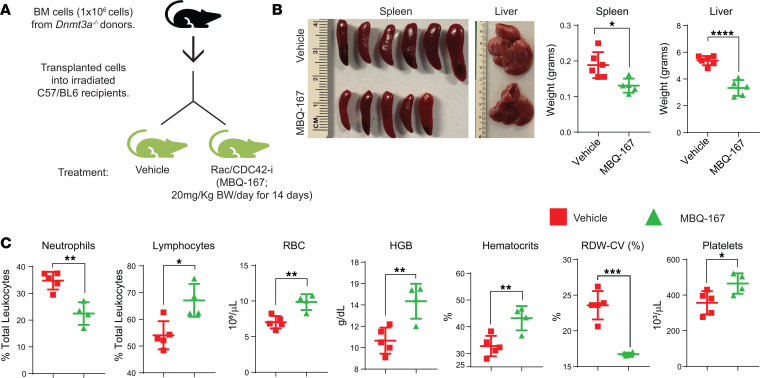
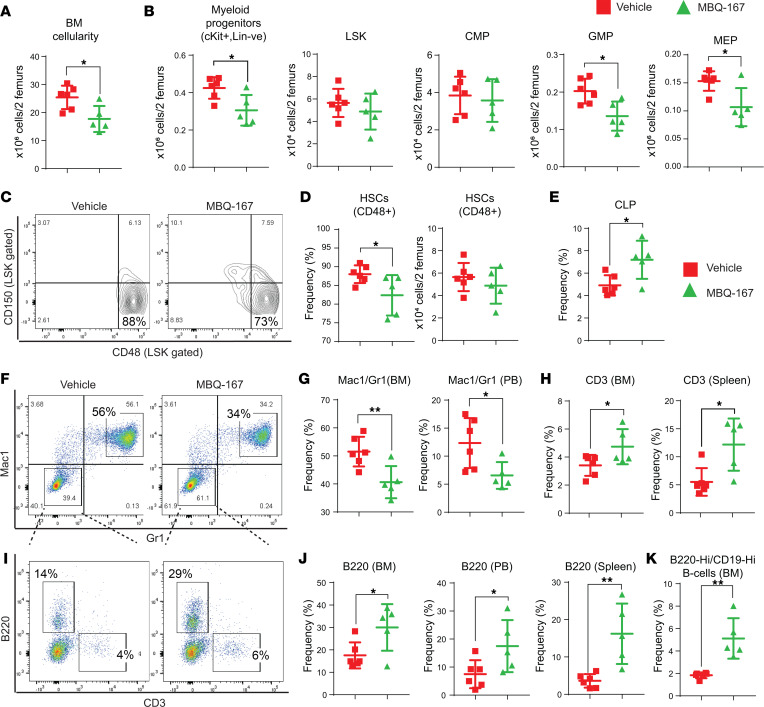
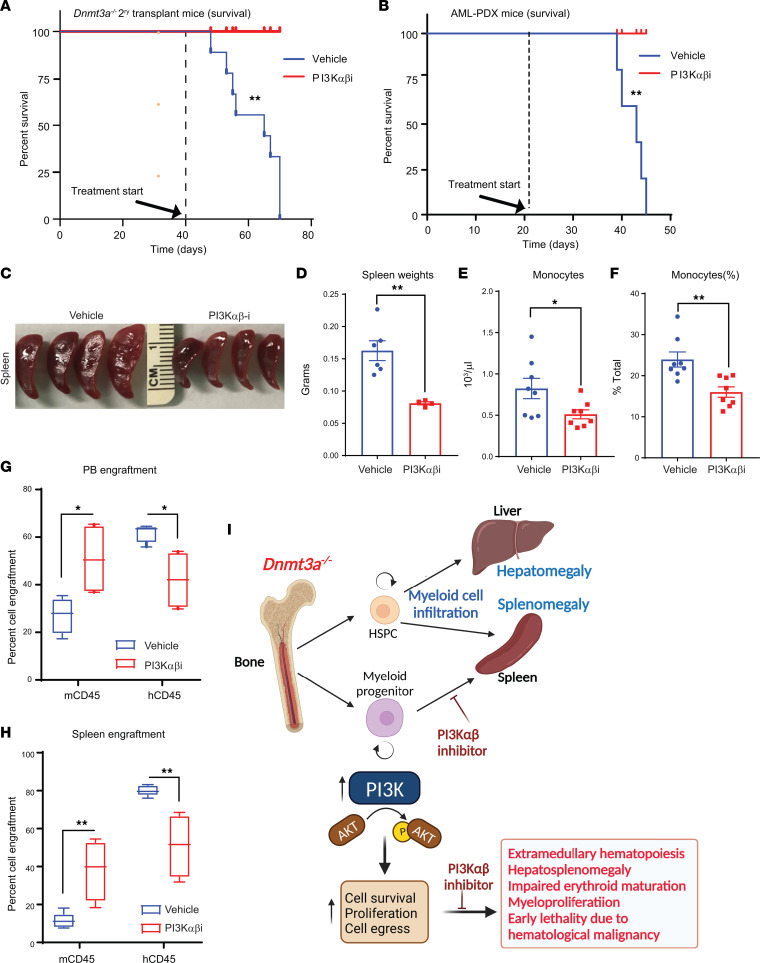
Loss-of-function mutations in the DNA methyltransferase 3A (DNMT3A) are seen in a large number of patients with acute myeloid leukemia (AML) with normal cytogenetics and are frequently associated with poor prognosis. DNMT3A mutations are an early preleukemic event, which - when combined with other genetic lesions - result in full-blown leukemia. Here, we show that loss of Dnmt3a in hematopoietic stem and progenitor cells (HSC/Ps) results in myeloproliferation, which is associated with hyperactivation of the phosphatidylinositol 3-kinase (PI3K) pathway. PI3Kα/β or the PI3Kα/δ inhibitor treatment partially corrects myeloproliferation, although the partial rescue is more efficient in response to the PI3Kα/β inhibitor treatment. In vivo RNA-Seq analysis on drug-treated Dnmt3a-/- HSC/Ps showed a reduction in the expression of genes associated with chemokines, inflammation, cell attachment, and extracellular matrix compared with controls. Remarkably, drug-treated leukemic mice showed a reversal in the enhanced fetal liver HSC-like gene signature observed in vehicle-treated Dnmt3a-/- LSK cells as well as a reduction in the expression of genes involved in regulating actin cytoskeleton-based functions, including the RHO/RAC GTPases. In a human PDX model bearing DNMT3A mutant AML, PI3Kα/β inhibitor treatment prolonged their survival and rescued the leukemic burden. Our results identify a potentially new target for treating DNMT3A mutation-driven myeloid malignancies.
Keywords: Hematology; Hematopoietic stem cells; Leukemias.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
