

Integrating Hydrogen Deuterium Exchange-Mass Spectrometry with Molecular Simulations Enables Quantification of the Conformational Populations of the Sugar Transporter XylE
- PMID: 36976935
- PMCID: PMC10103171
- DOI: 10.1021/jacs.2c06148
Integrating Hydrogen Deuterium Exchange-Mass Spectrometry with Molecular Simulations Enables Quantification of the Conformational Populations of the Sugar Transporter XylE
Abstract
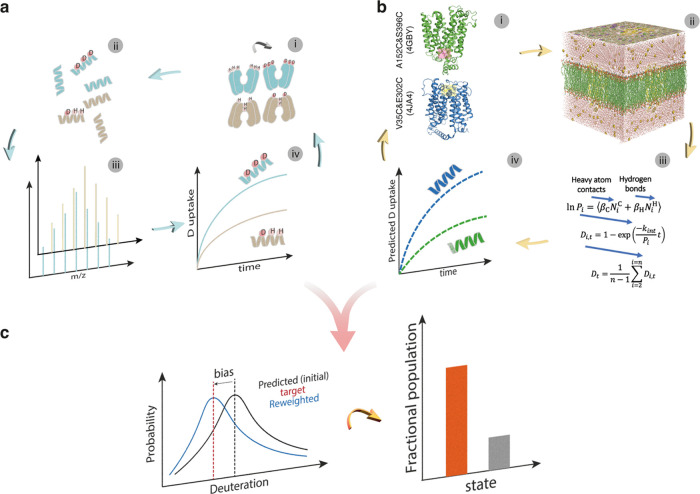
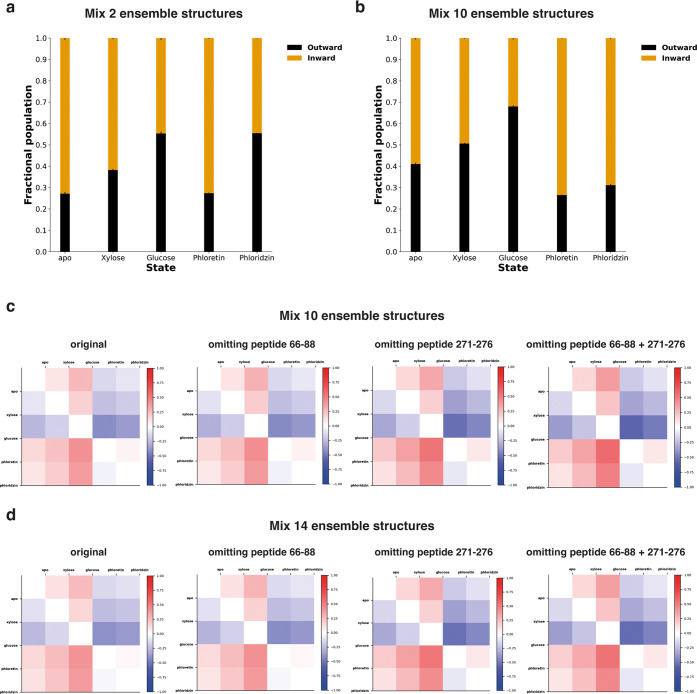
A yet unresolved challenge in structural biology is to quantify the conformational states of proteins underpinning function. This challenge is particularly acute for membrane proteins owing to the difficulties in stabilizing them for in vitro studies. To address this challenge, we present an integrative strategy that combines hydrogen deuterium exchange-mass spectrometry (HDX-MS) with ensemble modeling. We benchmark our strategy on wild-type and mutant conformers of XylE, a prototypical member of the ubiquitous Major Facilitator Superfamily (MFS) of transporters. Next, we apply our strategy to quantify conformational ensembles of XylE embedded in different lipid environments. Further application of our integrative strategy to substrate-bound and inhibitor-bound ensembles allowed us to unravel protein-ligand interactions contributing to the alternating access mechanism of secondary transport in atomistic detail. Overall, our study highlights the potential of integrative HDX-MS modeling to capture, accurately quantify, and subsequently visualize co-populated states of membrane proteins in association with mutations and diverse substrates and inhibitors.
Conflict of interest statement
The authors declare no competing financial interest.
Figures




References
-
- Trabjerg E.; Nazari Z. E.; Rand K. D. Conformational analysis of complex protein states by hydrogen/deuterium exchange mass spectrometry (HDX-MS): Challenges and emerging solutions. TrAC, Trends Anal. Chem. 2018, 106, 125–138. 10.1016/j.trac.2018.06.008. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
