

SARS-CoV2 billion-compound docking
- PMID: 36977690
- PMCID: PMC10044124
- DOI: 10.1038/s41597-023-01984-9
SARS-CoV2 billion-compound docking
Abstract
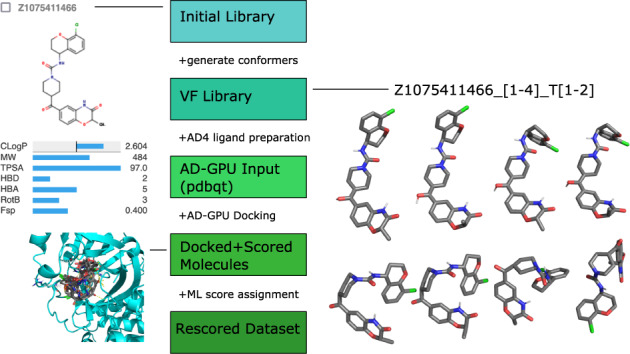
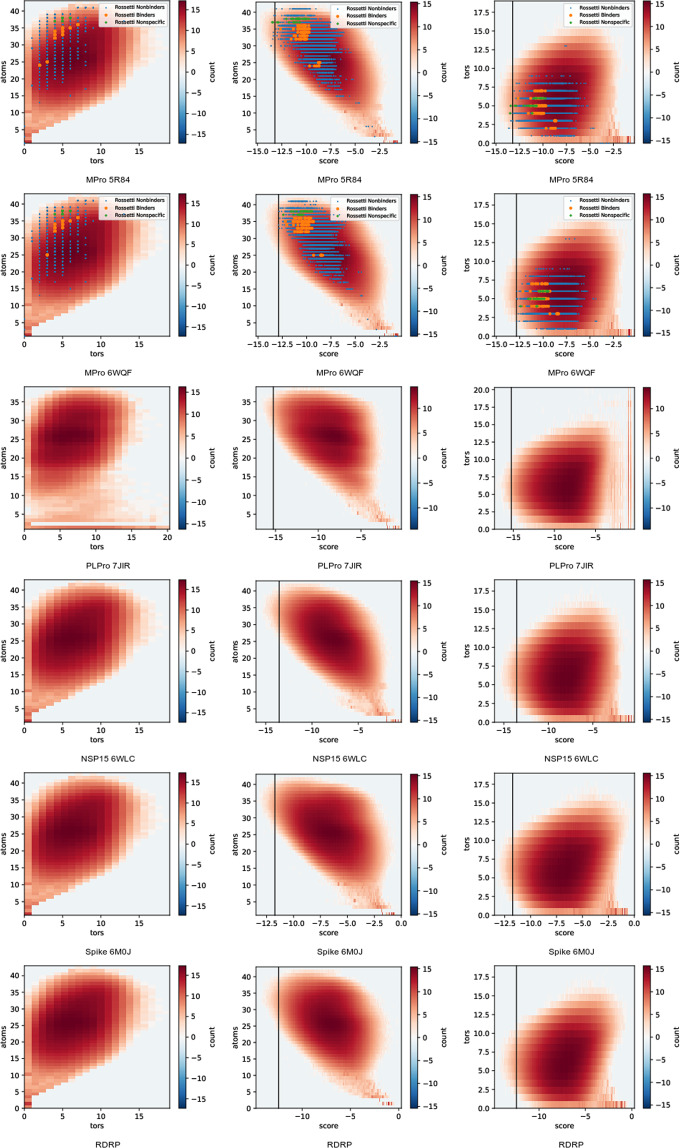
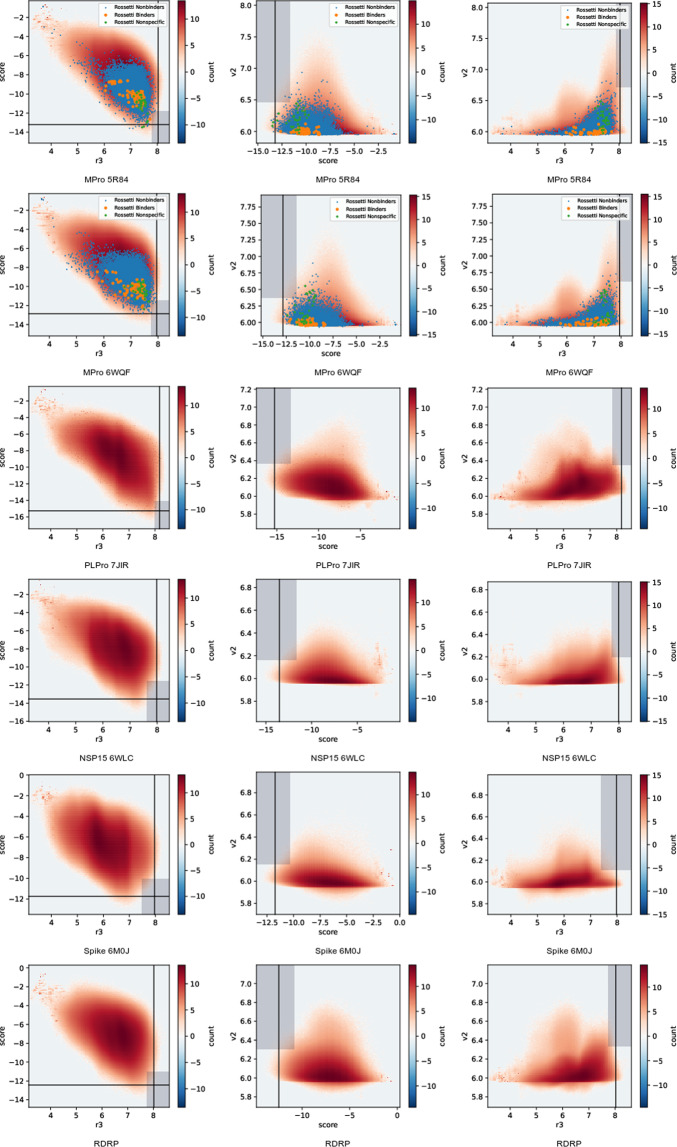
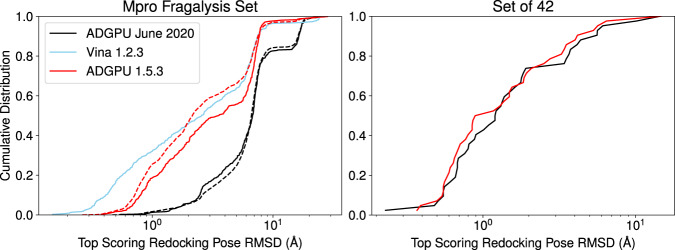
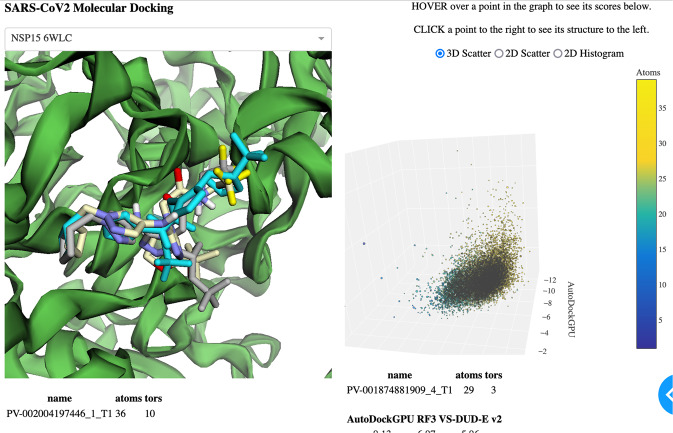
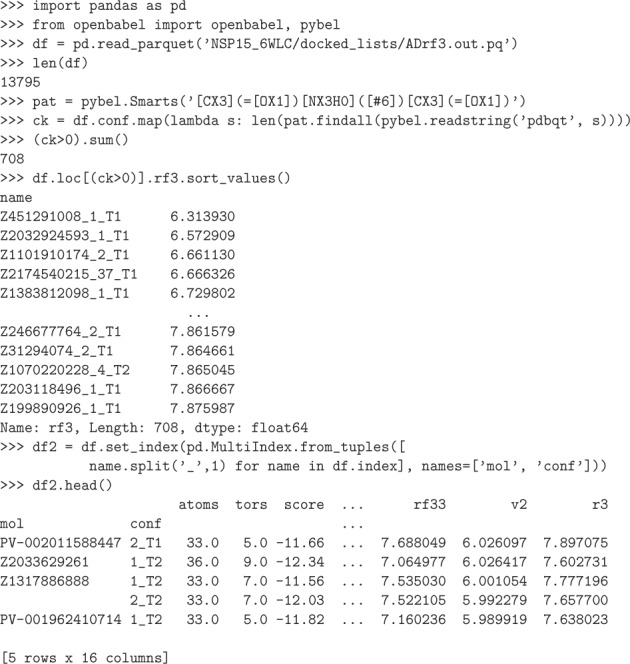
This dataset contains ligand conformations and docking scores for 1.4 billion molecules docked against 6 structural targets from SARS-CoV2, representing 5 unique proteins: MPro, NSP15, PLPro, RDRP, and the Spike protein. Docking was carried out using the AutoDock-GPU platform on the Summit supercomputer and Google Cloud. The docking procedure employed the Solis Wets search method to generate 20 independent ligand binding poses per compound. Each compound geometry was scored using the AutoDock free energy estimate, and rescored using RFScore v3 and DUD-E machine-learned rescoring models. Input protein structures are included, suitable for use by AutoDock-GPU and other docking programs. As the result of an exceptionally large docking campaign, this dataset represents a valuable resource for discovering trends across small molecule and protein binding sites, training AI models, and comparing to inhibitor compounds targeting SARS-CoV-2. The work also gives an example of how to organize and process data from ultra-large docking screens.
© 2023. UT-Battelle, LLC.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Singh, S., Bani Baker, Q. & Singh, D. B. Molecular docking and molecular dynamics simulation. In Singh, D. B. & Pathak, R. K. (eds.) Bioinformatics, chap. 18, 291–304, 10.1016/B978-0-323-89775-4.00014-6 (Academic Press, 2022).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
