

Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries
- PMID: 36978796
- PMCID: PMC10045332
- DOI: 10.3390/antiox12030548
Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries
Abstract
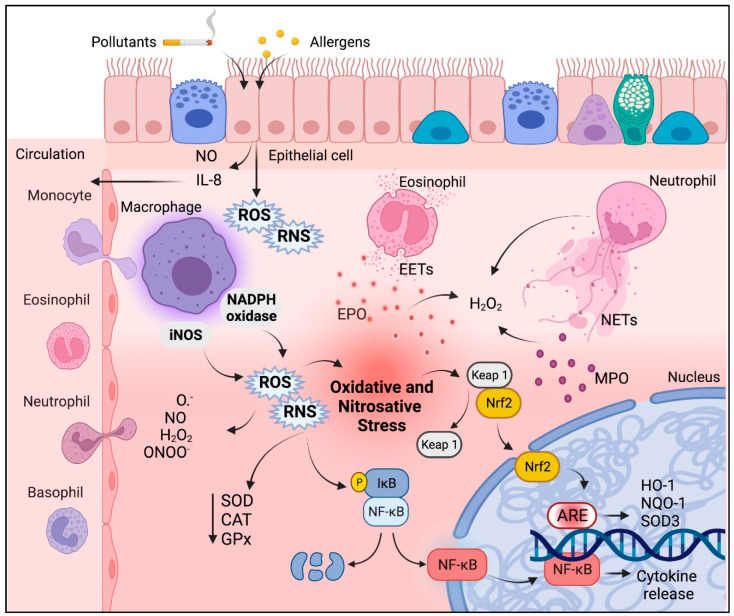
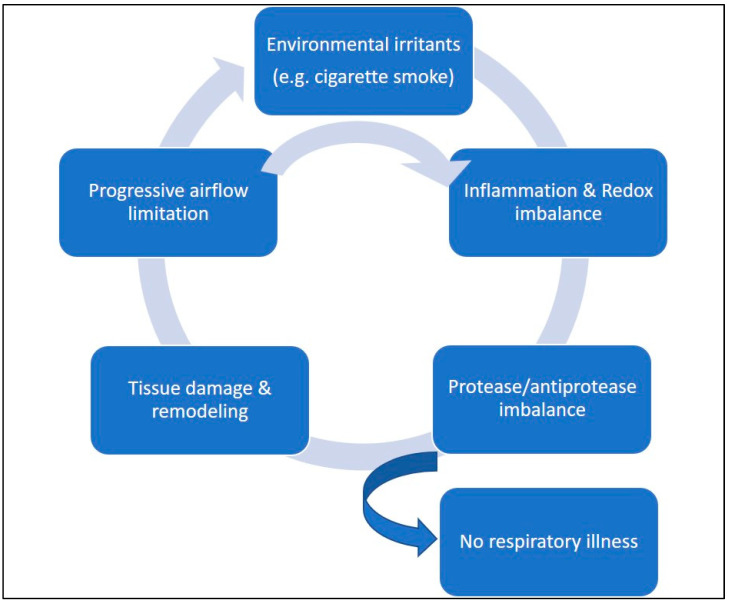
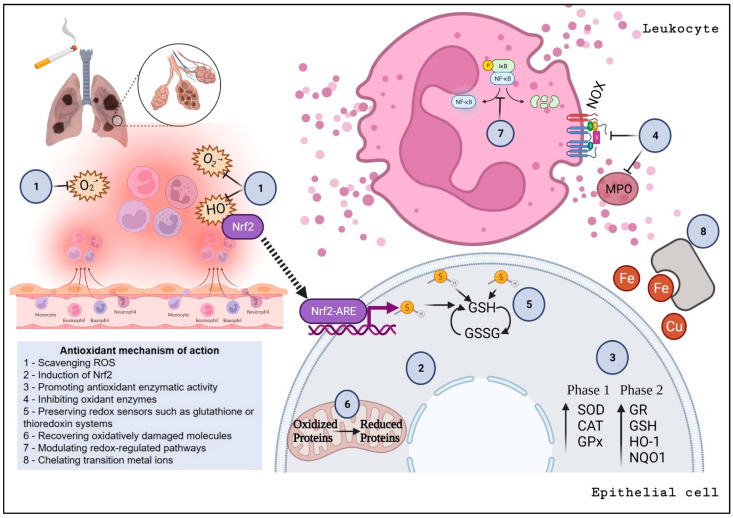
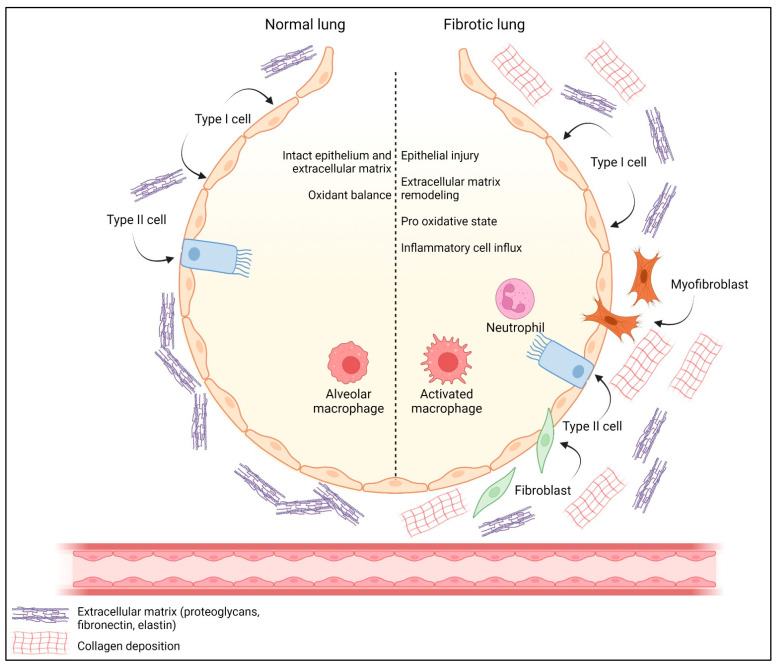
Acute and chronic lung injuries are among the leading causes of mortality worldwide. Lung injury can affect several components of the respiratory system, including the airways, parenchyma, and pulmonary vasculature. Although acute and chronic lung injuries represent an enormous economic and clinical burden, currently available therapies primarily focus on alleviating disease symptoms rather than reversing and/or preventing lung pathology. Moreover, some supportive interventions, such as oxygen and mechanical ventilation, can lead to (further) deterioration of lung function and even the development of permanent injuries. Lastly, sepsis, which can originate extrapulmonary or in the respiratory system itself, contributes to many cases of lung-associated deaths. Considering these challenges, we aim to summarize molecular and cellular mechanisms, with a particular focus on airway inflammation and oxidative stress that lead to the characteristic pathophysiology of acute and chronic lung injuries. In addition, we will highlight the limitations of current therapeutic strategies and explore new antioxidant-based drug options that could potentially be effective in managing acute and chronic lung injuries.
Keywords: asthma; chronic obstructive pulmonary disease; emphysema; hyperoxia; pulmonary fibrosis; sepsis; ventilator-induced lung injury.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Bousquet J., Kiley J., Bateman E.D., Viegi G., Cruz A.A., Khaltaev N., Ait Khaled N., Baena-Cagnani C.E., Barreto M.L., Billo N., et al. Prioritised research agenda for prevention and control of chronic respiratory diseases. Eur. Respir. J. 2010;36:995–1001. doi: 10.1183/09031936.00012610. - DOI - PubMed
-
- Andersen Z.J., Hoffmann B., Morawska L., Adams M., Furman E., Yorgancioglu A., Greenbaum D., Neira M., Brunekreef B., Forastiere F., et al. Air pollution and COVID-19: Clearing the air and charting a post-pandemic course: A joint workshop report of ERS, ISEE, HEI and WHO. Eur. Respir. J. 2021;58:2101063. doi: 10.1183/13993003.01063-2021. - DOI - PMC - PubMed
-
- Lammi M.R., Baughman R.P., Birring S.S., Russell A.M., Ryu J.H., Scholand M., Distler O., LeSage D., Sarver C., Antoniou K., et al. Outcome Measures for Clinical Trials in Interstitial Lung Diseases. Curr. Respir. Med. Rev. 2015;11:163–174. doi: 10.2174/1573398X11666150619183527. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
