

Evaluation of Human-Induced Pluripotent Stem Cells Derived from a Patient with Schwartz-Jampel Syndrome Revealed Distinct Hyperexcitability in the Skeletal Muscles
- PMID: 36979792
- PMCID: PMC10045278
- DOI: 10.3390/biomedicines11030814
Evaluation of Human-Induced Pluripotent Stem Cells Derived from a Patient with Schwartz-Jampel Syndrome Revealed Distinct Hyperexcitability in the Skeletal Muscles
Abstract
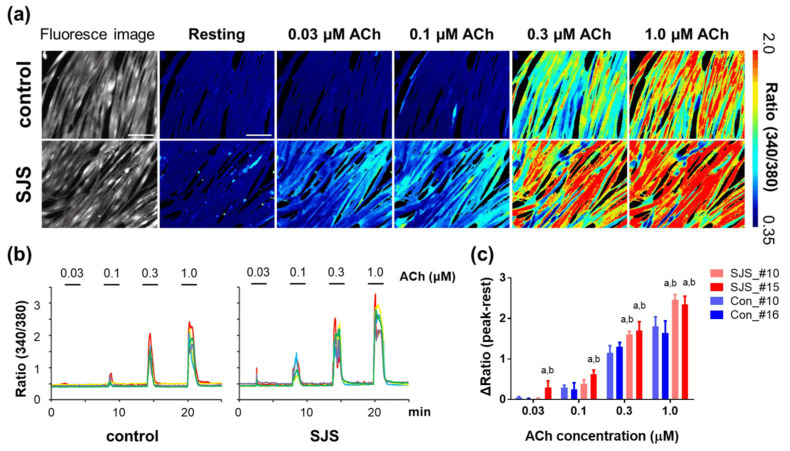
Schwartz-Jampel syndrome (SJS) is an autosomal recessive disorder caused by loss-of-function mutations in heparan sulfate proteoglycan 2 (HSPG2), which encodes the core basement membrane protein perlecan. Myotonia is a major criterion for the diagnosis of SJS; however, its evaluation is based solely on physical examination and can be challenging in neonates and young children. Furthermore, the pathomechanism underlying SJS-related myotonia is not fully understood, and effective treatments for SJS are limited. Here, we established a cellular model of SJS using patient-derived human-induced pluripotent stem cells. This model exhibited hyper-responsiveness to acetylcholine as a result of abnormalities in the perlecan molecule, which were confirmed via comparison of their calcium imaging with calcium imaging of satellite cells derived from Hspg2-/--Tg mice, which exhibit myotonic symptoms similar to SJS symptoms. Therefore, our results confirm the utility of creating cellular models for investigating SJS and their application in evaluating myotonia in clinical cases, while also providing a useful tool for the future screening of SJS therapies.
Keywords: Schwartz–Jampel syndrome; calcium imaging; human-induced pluripotent stem cell; myotonia; perlecan; skeletal muscle.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
-
- Nicole S., Davoine C.S., Topaloglu H., Cattolico L., Barral D., Beighton P., Hamida C.B., Hammouda H., Cruaud C., White P.S., et al. Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia) Nat. Genet. 2000;26:480–483. doi: 10.1038/82638. - DOI - PubMed
-
- Dagoneau N., Scheffer D., Huber C., Al-Gazali L.I., Di Rocco M., Godard A., Martinovic J., Raas-Rothschild A., Sigaudy S., Unger S., et al. Null leukemia inhibitory factor receptor (LIFR) mutations in Stuve-Wiedemann/Schwartz-Jampel type 2 syndrome. Am. J. Hum. Genet. 2004;74:298–305. doi: 10.1086/381715. - DOI - PMC - PubMed
-
- Arikawa-Hirasawa E., Le A.H., Nishino I., Nonaka I., Ho N.C., Francomano C.A., Govindraj P., Hassell J.R., Devaney J.M., Spranger J., et al. Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia. Am. J. Hum. Genet. 2002;70:1368–1375. doi: 10.1086/340390. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
