

Co-Occurring X-Linked Agammaglobulinemia and X-Linked Chronic Granulomatous Disease: Two Isolated Pathogenic Variants in One Patient
- PMID: 36979938
- PMCID: PMC10046124
- DOI: 10.3390/biomedicines11030959
Co-Occurring X-Linked Agammaglobulinemia and X-Linked Chronic Granulomatous Disease: Two Isolated Pathogenic Variants in One Patient
Abstract
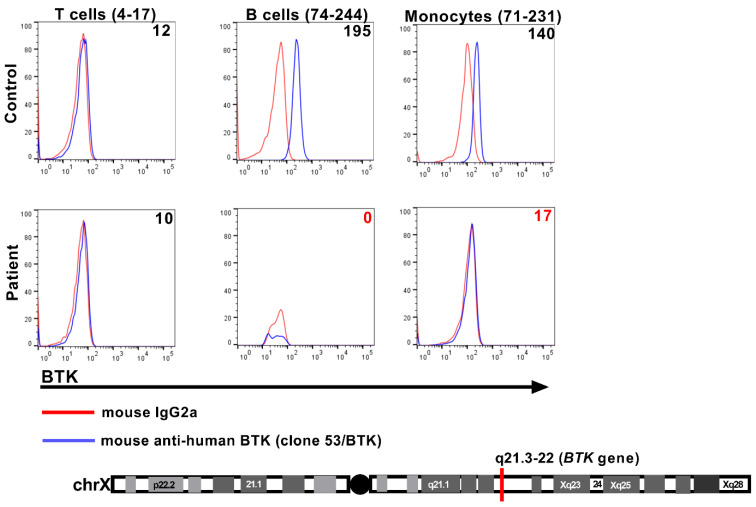
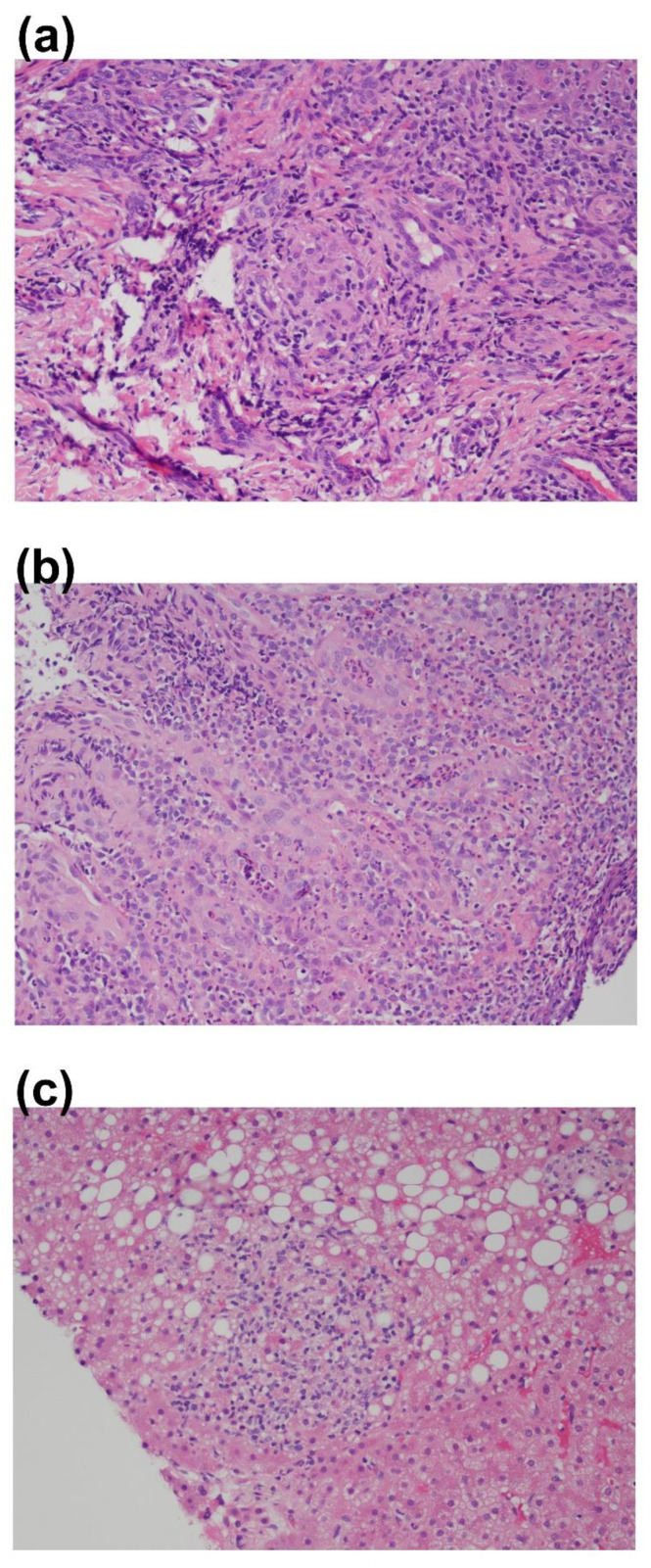
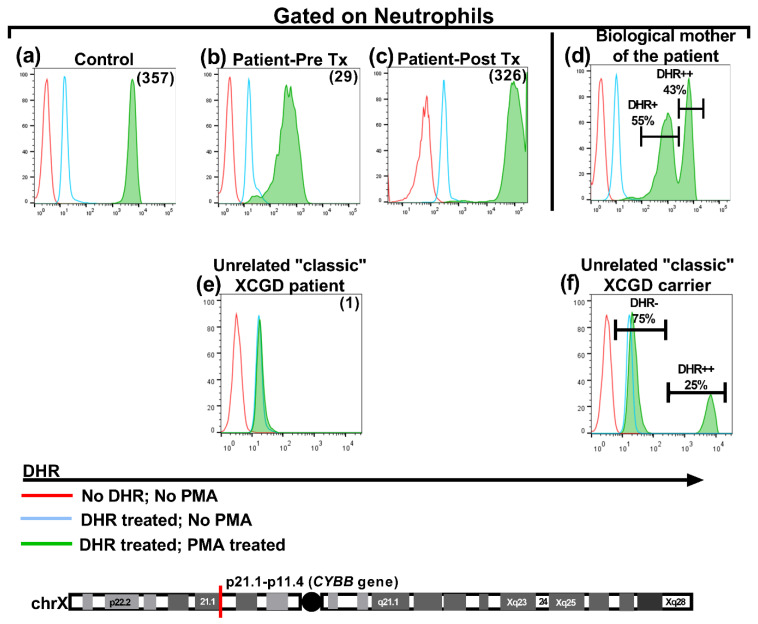
We present a unique and unusual case of a male patient diagnosed with two coexisting and typically unassociated X-linked conditions: he was initially diagnosed with X-linked agammaglobulinemia (XLA) followed by a diagnosis of X-linked chronic granulomatous disease (XCGD) and an as of yet unpublished hypomorphic gp91phox variant in the CYBB gene. The latter was tested after the finding of granulomatous gingivitis. Hematopoietic stem cell transplant (HSCT) was performed due to severe colitis and nodular regenerative hyperplasia (NRH) of the liver. Following transplant, complete donor engraftment was observed with the restoration of a normal oxidative burst and full restoration of normal levels of circulating, mature CD19+ B cells. This case is singular in that it does not involve a contiguous gene syndrome in which deleted genes are in close proximity to either BTK and CYBB, which has been previously reported. To our knowledge, this is the first reported case of XLA and XCGD co-existing in a single patient and of having both inborn errors of immunity successfully treated by HSCT.
Keywords: BTK; HSCT; X-linked agammaglobulinemia; X-linked chronic granulomatous disease; XLA; multi-genetic diagnosis.
Conflict of interest statement
The authors do not have any related conflict of interest.
Figures
References
-
- Chan K.-W., Wong C.-Y., Leung D., Yang X., Fok S.F.S., Mak P.H.S., Yao L., Ma W., Mao H., Zhao X., et al. Targeted Gene Sanger Sequencing Should Remain the First-Tier Genetic Test for Children Suspected to Have the Five Common X-Linked Inborn Errors of Immunity. Front. Immunol. 2022;13:883446. doi: 10.3389/fimmu.2022.883446. - DOI - PMC - PubMed
-
- El-Sayed Z.A., Abramova I., Aldave J.C., Al-Herz W., Bezrodnik L., Boukari R., Bousfiha A.A., Cancrini C., Condino-Neto A., Dbaibo G., et al. X-linked agammaglobulinemia (XLA): Phenotype, diagnosis, and therapeutic challenges around the world. World Allergy Organ. J. 2019;12:100018. doi: 10.1016/j.waojou.2019.100018. - DOI - PMC - PubMed
-
- O’Toole D., Groth D., Wright H., Bonilla F.A., Fuleihan R.L., Cunningham-Rundles C., Sullivan K.E., Ochs H.D., Marsh R., Feuille E. X-Linked Agammaglobulinemia: Infection Frequency and Infection-Related Mortality in the USIDNET Registry. J. Clin. Immunol. 2022;42:827–836. doi: 10.1007/s10875-022-01237-1. - DOI - PMC - PubMed
-
- Marciano B.E., Spalding C., Fitzgerald A., Mann D., Brown T., Osgood S., Yockey L., Darnell D.N., Barnhart L., Daub J., et al. Common severe infections in chronic granulomatous disease. Clinical infectious diseases: An official publication of the Infectious Diseases Society of America. Clinical Infectious Diseases. 2015;60:1176–1183. doi: 10.1093/cid/ciu1154. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
