

A Comprehensive and Integrative Approach to MeCP2 Disease Transcriptomics
- PMID: 36982190
- PMCID: PMC10049497
- DOI: 10.3390/ijms24065122
A Comprehensive and Integrative Approach to MeCP2 Disease Transcriptomics
Abstract
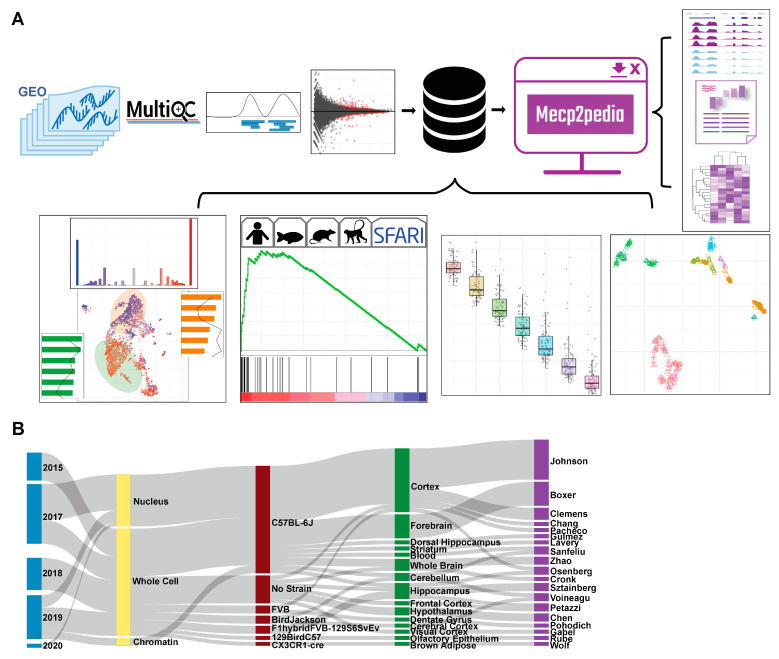
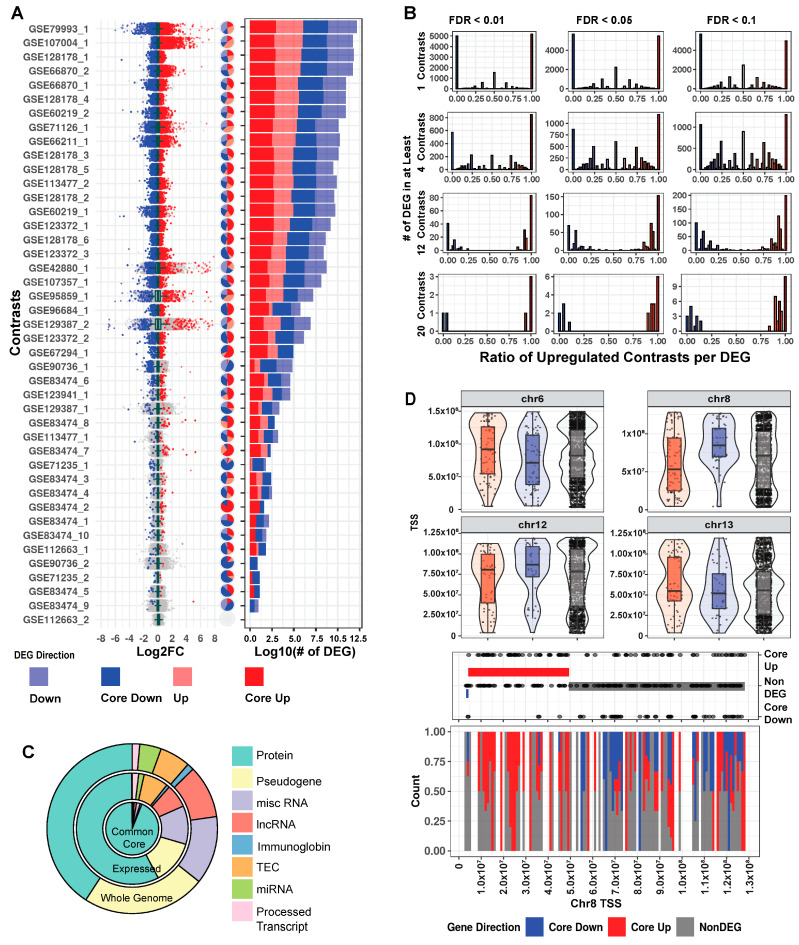
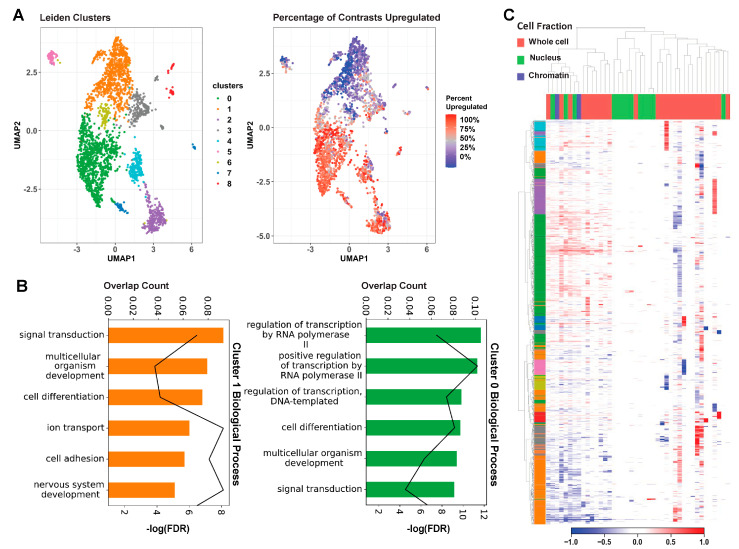
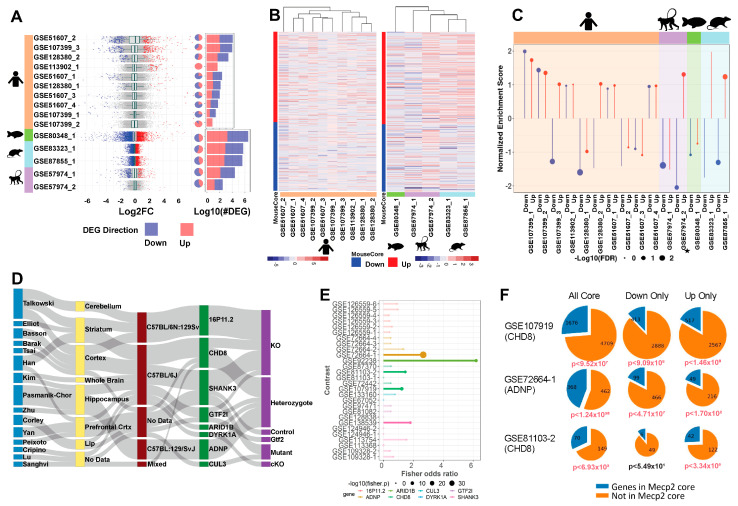
Mutations in MeCP2 result in a crippling neurological disease, but we lack a lucid picture of MeCP2's molecular role. Individual transcriptomic studies yield inconsistent differentially expressed genes. To overcome these issues, we demonstrate a methodology to analyze all modern public data. We obtained relevant raw public transcriptomic data from GEO and ENA, then homogeneously processed it (QC, alignment to reference, differential expression analysis). We present a web portal to interactively access the mouse data, and we discovered a commonly perturbed core set of genes that transcends the limitations of any individual study. We then found functionally distinct, consistently up- and downregulated subsets within these genes and some bias to their location. We present this common core of genes as well as focused cores for up, down, cell fraction models, and some tissues. We observed enrichment for this mouse core in other species MeCP2 models and observed overlap with ASD models. By integrating and examining transcriptomic data at scale, we have uncovered the true picture of this dysregulation. The vast scale of these data enables us to analyze signal-to-noise, evaluate a molecular signature in an unbiased manner, and demonstrate a framework for future disease focused informatics work.
Keywords: MeCP2; MeCP2 duplication syndrome; RNA-seq; Rett syndrome; data portal; differential expression analysis; meta-analysis; mouse models.
Conflict of interest statement
The authors declare no competing interests.
Figures
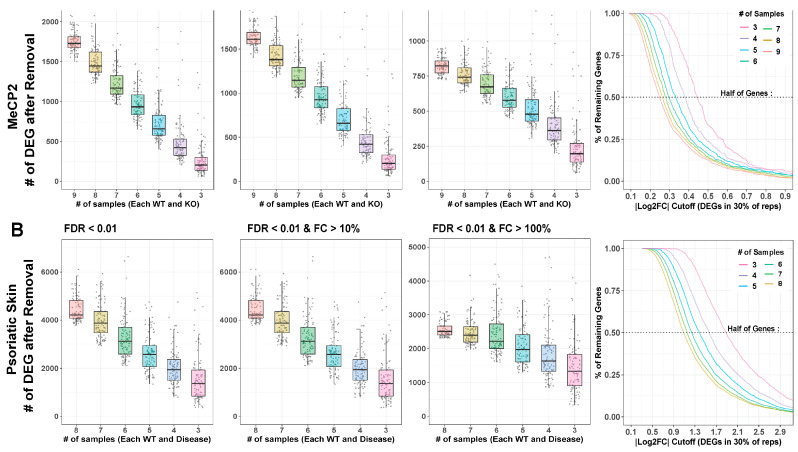
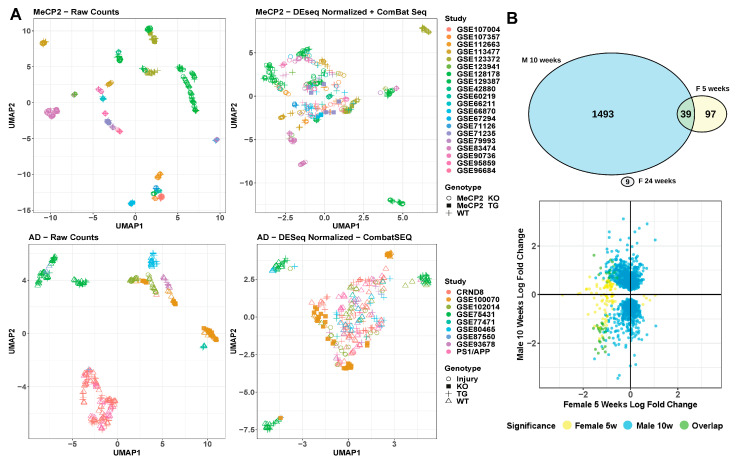
References
-
- Smith J.R., Hayman G.T., Wang S.J., Laulederkind S., Hoffman M.J., Kaldunski M.L., Tutaj M., Thota J., Nalabolu H.S., Ellanki S., et al. The Year of the Rat: The Rat Genome Database at 20: A multi-species knowledgebase and analysis platform. Nucleic Acids Res. 2020;48:D731–D742. doi: 10.1093/nar/gkz1041. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
