

Pharmacological Chaperones and Protein Conformational Diseases: Approaches of Computational Structural Biology
- PMID: 36982893
- PMCID: PMC10054308
- DOI: 10.3390/ijms24065819
Pharmacological Chaperones and Protein Conformational Diseases: Approaches of Computational Structural Biology
Abstract
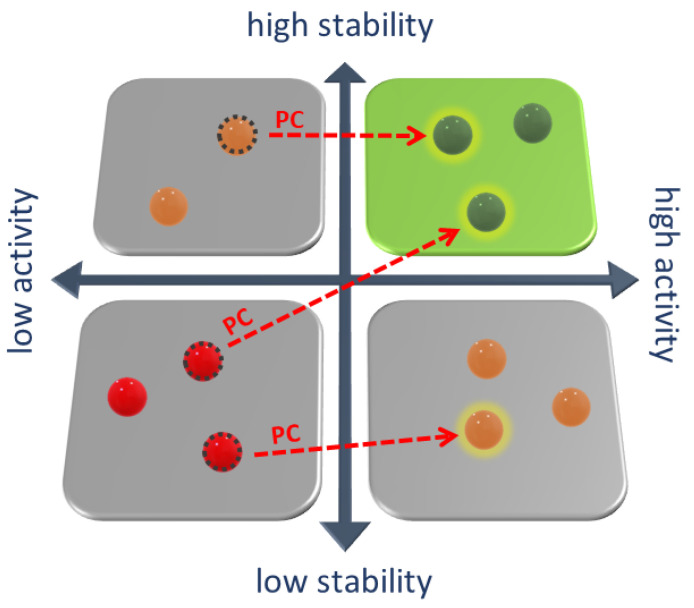
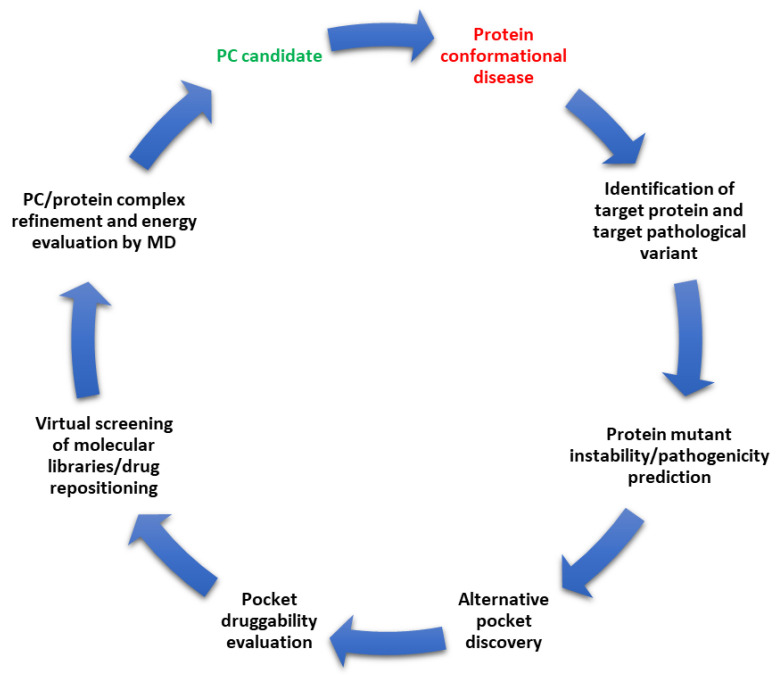
Whenever a protein fails to fold into its native structure, a profound detrimental effect is likely to occur, and a disease is often developed. Protein conformational disorders arise when proteins adopt abnormal conformations due to a pathological gene variant that turns into gain/loss of function or improper localization/degradation. Pharmacological chaperones are small molecules restoring the correct folding of a protein suitable for treating conformational diseases. Small molecules like these bind poorly folded proteins similarly to physiological chaperones, bridging non-covalent interactions (hydrogen bonds, electrostatic interactions, and van der Waals contacts) loosened or lost due to mutations. Pharmacological chaperone development involves, among other things, structural biology investigation of the target protein and its misfolding and refolding. Such research can take advantage of computational methods at many stages. Here, we present an up-to-date review of the computational structural biology tools and approaches regarding protein stability evaluation, binding pocket discovery and druggability, drug repurposing, and virtual ligand screening. The tools are presented as organized in an ideal workflow oriented at pharmacological chaperones' rational design, also with the treatment of rare diseases in mind.
Keywords: computational structural biology; drug repurposing; molecular docking; pharmacological chaperones; pocket druggability; protein conformational diseases; protein stability; transient pockets; virtual screening.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Screening methods for identifying pharmacological chaperones.Mol Biosyst. 2017 Mar 28;13(4):638-647. doi: 10.1039/c6mb00866f. Mol Biosyst. 2017. PMID: 28265599 Review.
-
Computational methods to assist in the discovery of pharmacological chaperones for rare diseases.Brief Bioinform. 2022 Sep 20;23(5):bbac198. doi: 10.1093/bib/bbac198. Brief Bioinform. 2022. PMID: 35595532 Review.
-
Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease.Traffic. 2004 Nov;5(11):821-37. doi: 10.1111/j.1600-0854.2004.00232.x. Traffic. 2004. PMID: 15479448 Review.
-
Second-Generation Pharmacological Chaperones: Beyond Inhibitors.Molecules. 2020 Jul 9;25(14):3145. doi: 10.3390/molecules25143145. Molecules. 2020. PMID: 32660097 Free PMC article. Review.
-
Role of misfolding in rare enzymatic deficits and use of pharmacological chaperones as therapeutic approach.Front Biosci (Landmark Ed). 2021 Dec 30;26(12):1627-1642. doi: 10.52586/5056. Front Biosci (Landmark Ed). 2021. PMID: 34994177 Review.
Cited by
-
Elucidating the Role of Wildtype and Variant FGFR2 Structural Dynamics in (Dys)Function and Disorder.Int J Mol Sci. 2024 Apr 20;25(8):4523. doi: 10.3390/ijms25084523. Int J Mol Sci. 2024. PMID: 38674107 Free PMC article.
-
Genetic variations in the IDUA gene in Tunisian MPS I families: Identification of a novel microdeletion disrupting substrate binding and structural insights.Mol Genet Metab Rep. 2025 Apr 19;43:101222. doi: 10.1016/j.ymgmr.2025.101222. eCollection 2025 Jun. Mol Genet Metab Rep. 2025. PMID: 40291162 Free PMC article.
-
Proteostasis regulation of GABAA receptors in neuronal function and disease.Biomed Pharmacother. 2025 May;186:117992. doi: 10.1016/j.biopha.2025.117992. Epub 2025 Mar 20. Biomed Pharmacother. 2025. PMID: 40112516 Free PMC article. Review.
-
Chronic Mexiletine Administration Increases Sodium Current in Non-Diseased Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes.Biomedicines. 2024 May 29;12(6):1212. doi: 10.3390/biomedicines12061212. Biomedicines. 2024. PMID: 38927420 Free PMC article.
-
Small-molecule correctors divert CFTR-F508del from ERAD by stabilizing sequential folding states.Mol Biol Cell. 2024 Feb 1;35(2):ar15. doi: 10.1091/mbc.E23-08-0336. Epub 2023 Nov 29. Mol Biol Cell. 2024. PMID: 38019608 Free PMC article.
References
-
- Reynaud E. Protein Misfolding and Degenerative Diseases. Nat. Educ. 2010;3:28.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
