

Metastasis in Neuroblastoma and Its Link to Autophagy
- PMID: 36983973
- PMCID: PMC10056181
- DOI: 10.3390/life13030818
Metastasis in Neuroblastoma and Its Link to Autophagy
Abstract
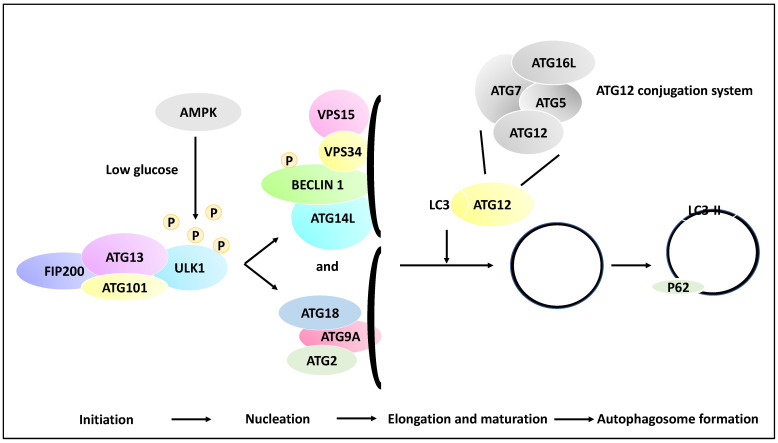
Neuroblastoma is a paediatric malignancy originating from the neural crest that commonly occurs in the abdomen and adrenal gland, leading to cancer-related deaths in children. Distant metastasis can be encountered at diagnosis in greater than half of these neuroblastoma patients. Autophagy, a self-degradative process, plays a key role in stress-related responses and the survival of cells and has been studied in neuroblastoma. Accordingly, in the early stages of metastasis, autophagy may suppress cancer cell invasion and migration, while its role may be reversed in later stages, and it may facilitate metastasis by enhancing cancer cell survival. To that end, a body of literature has revealed the mechanistic link between autophagy and metastasis in neuroblastoma in multiple steps of the metastatic cascade, including cancer cell invasion and migration, anoikis resistance, cancer cell dormancy, micrometastasis, and metastatic outbreak. This review aims to take a step forward and discuss the significance of multiple molecular players and compounds that may link autophagy to metastasis and map their function to various metastatic steps in neuroblastoma.
Keywords: autophagy; metastasis; neuroblastoma; paediatric cancers.
Conflict of interest statement
The author declares no conflict of interest.
Figures







References
-
- Cohn S.L., Pearson A.D.J., London W.B., Monclair T., Ambros P.F., Brodeur G.M., Faldum A., Hero B., Iehara T., Machin D., et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009;27:289–297. doi: 10.1200/JCO.2008.16.6785. - DOI - PMC - PubMed
-
- Brodeur G.M., Pritchard J., Berthold F., Carlsen N.L., Castel V., Castelberry R.P., De Bernardi B., Evans A.E., Favrot M., Hedborg F. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 1993;11:1466–1477. doi: 10.1200/JCO.1993.11.8.1466. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
