

Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease
- PMID: 36984562
- PMCID: PMC10059871
- DOI: 10.3390/medicina59030561
Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease
Abstract
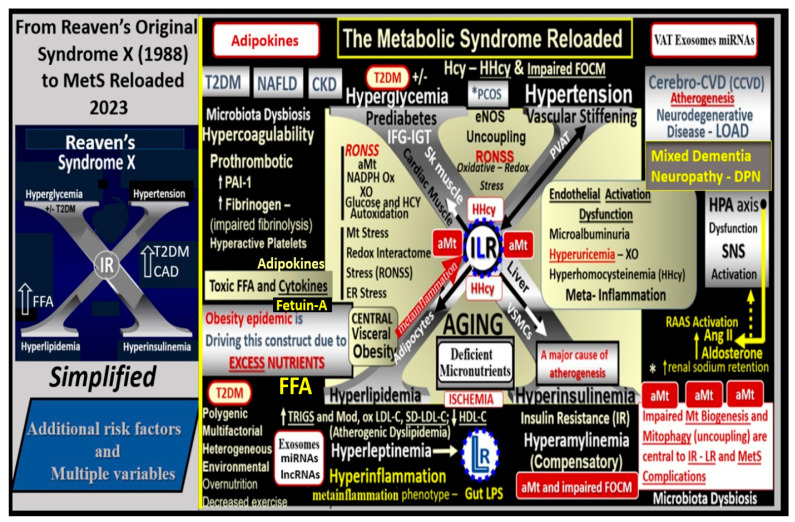
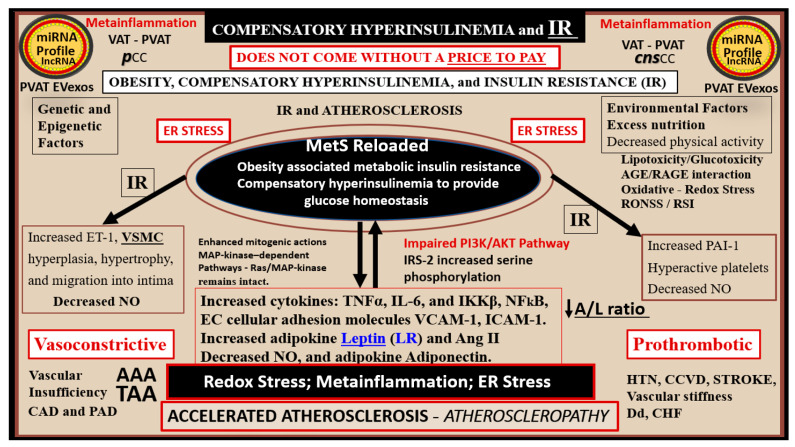
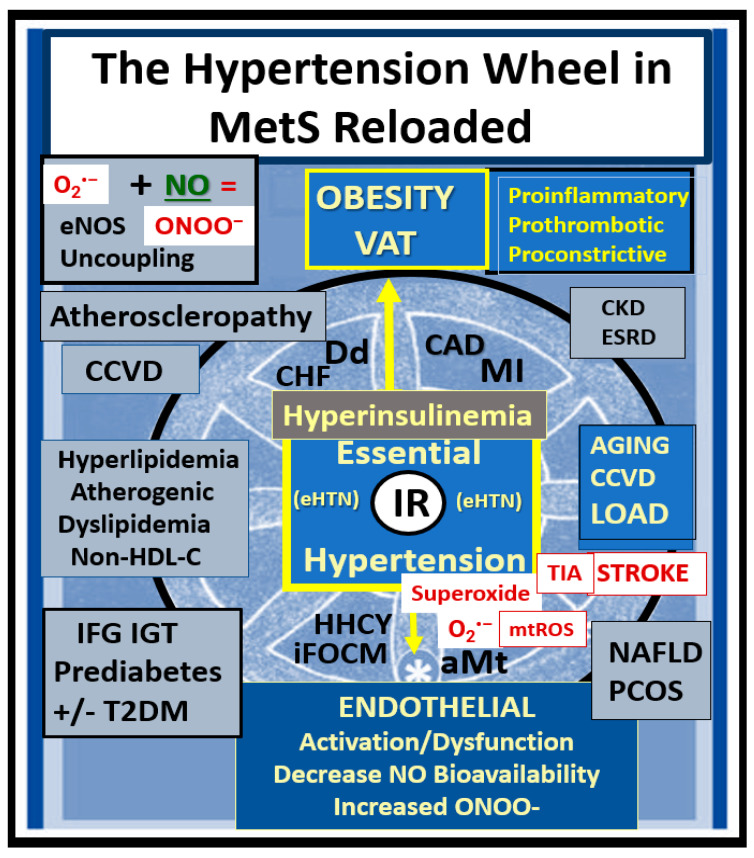
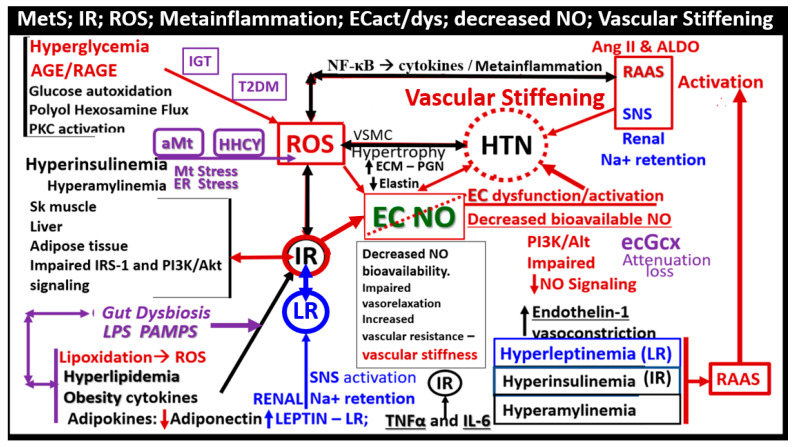
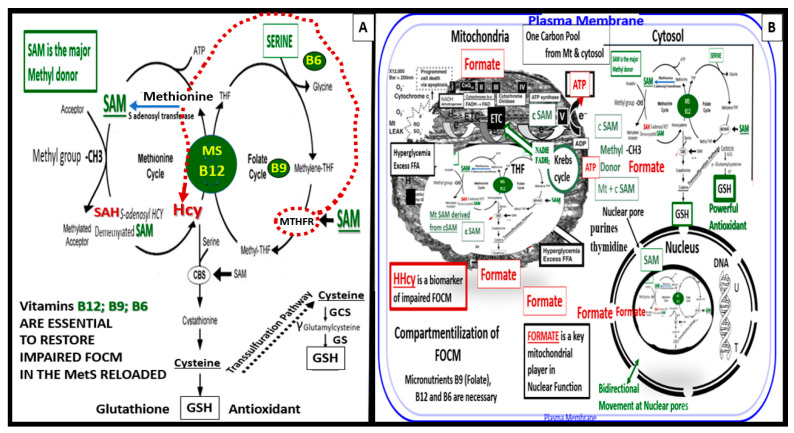
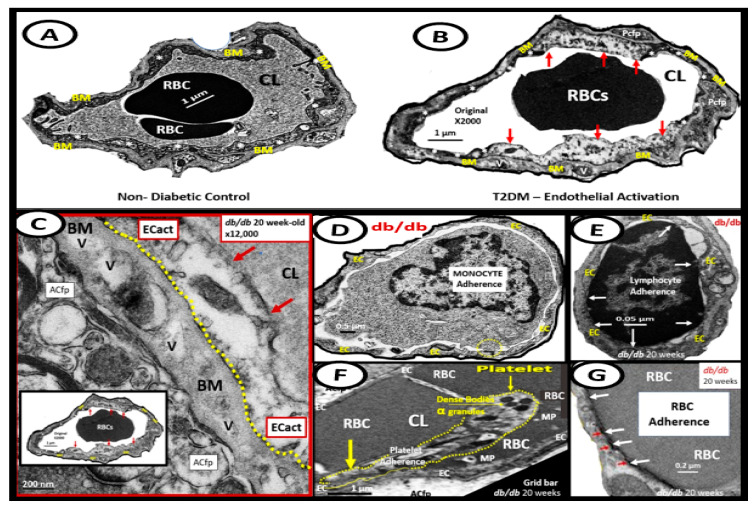
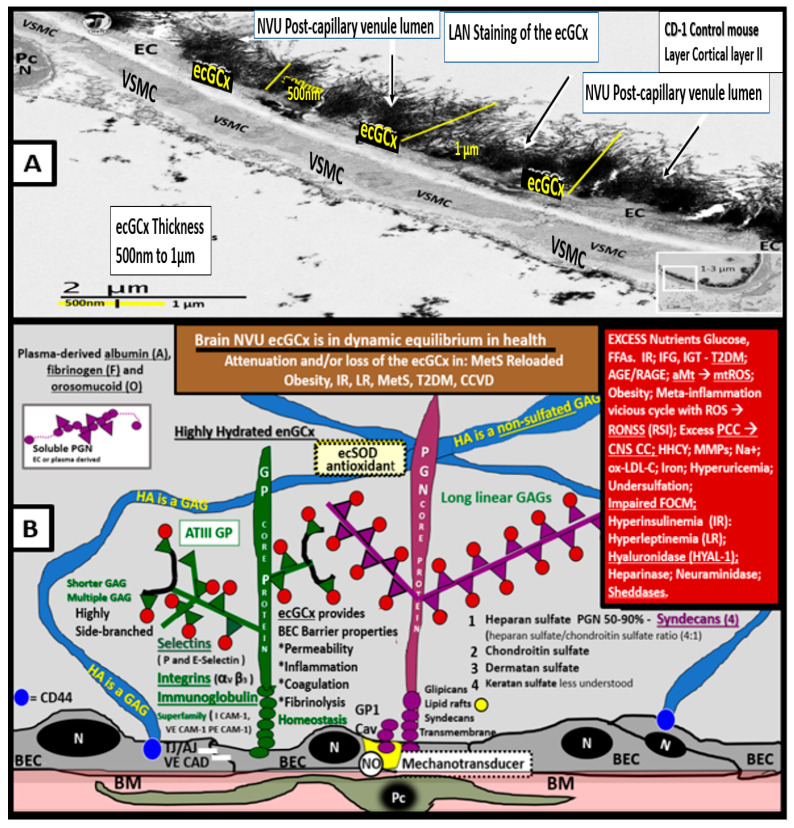
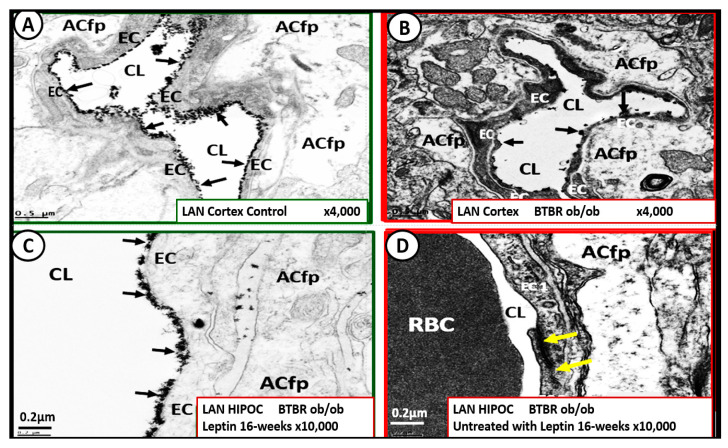
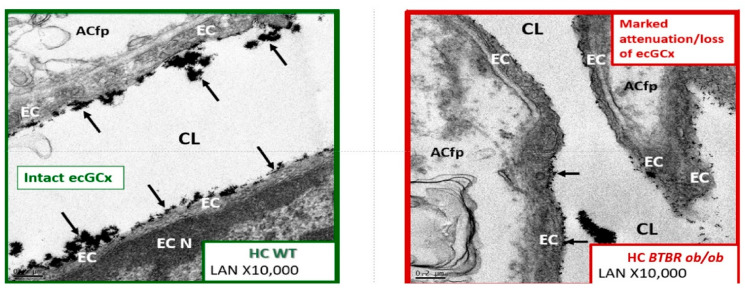
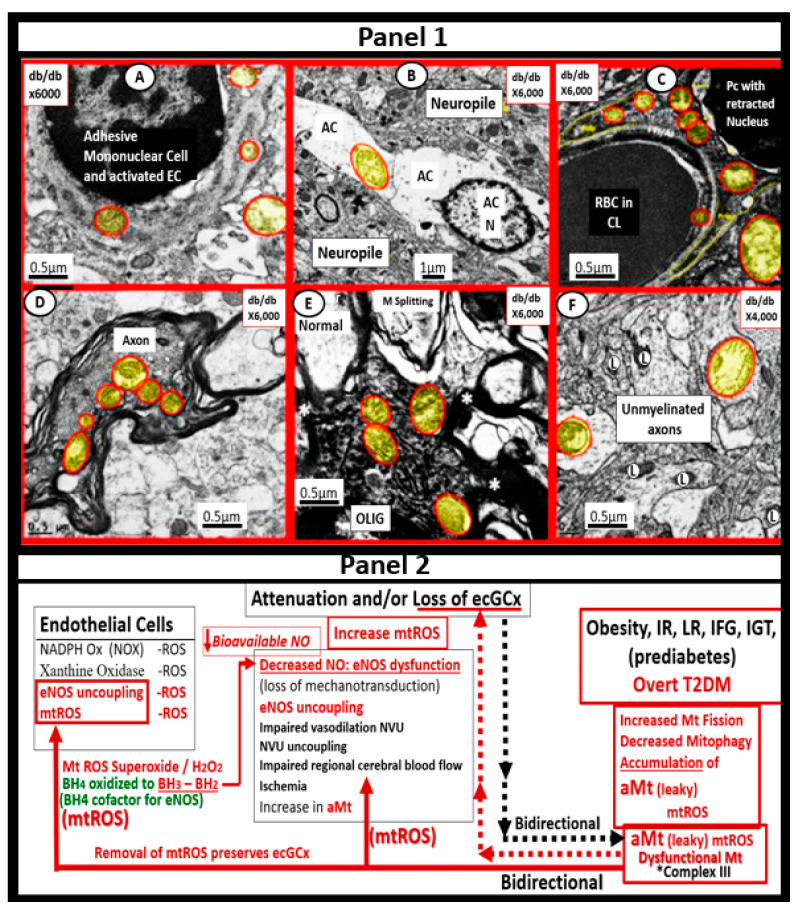
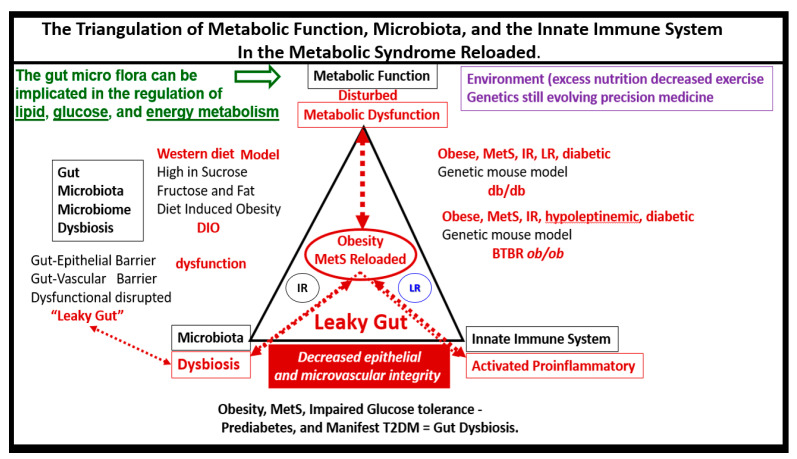
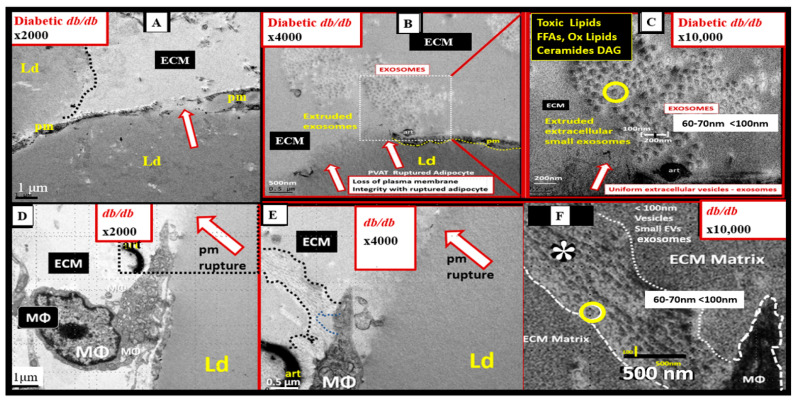
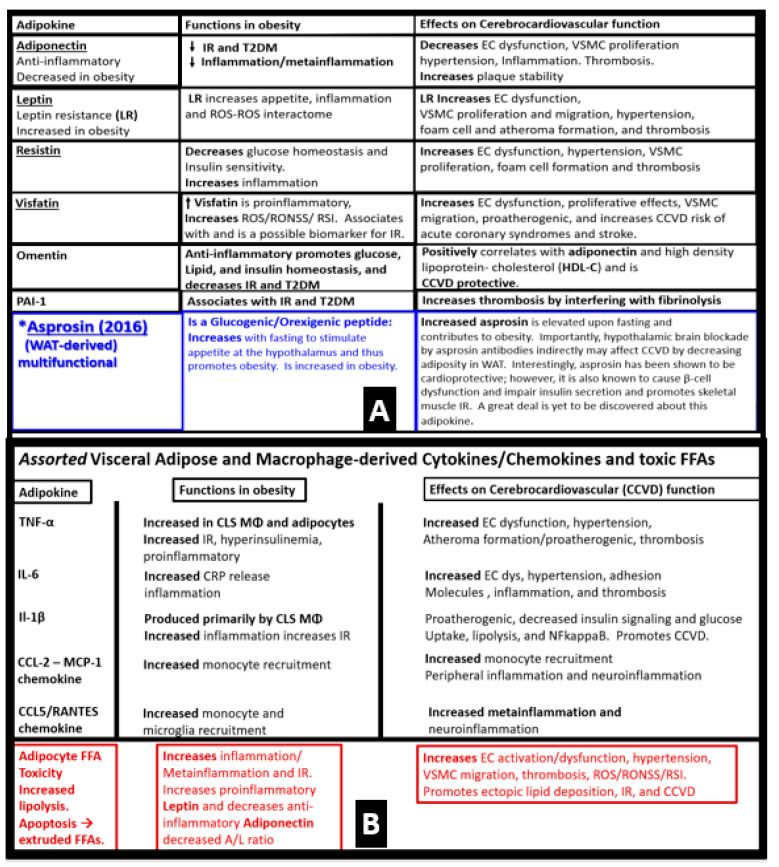
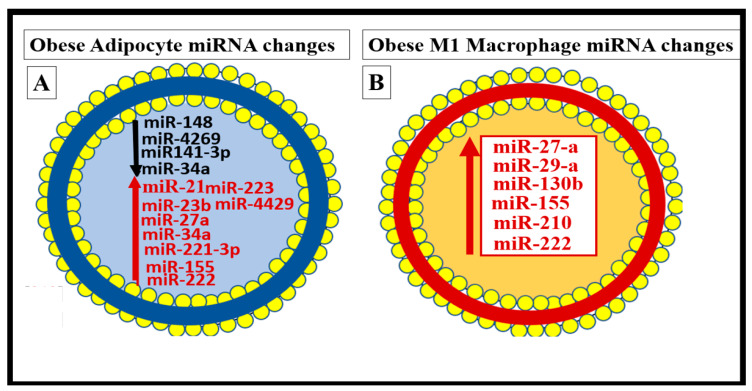
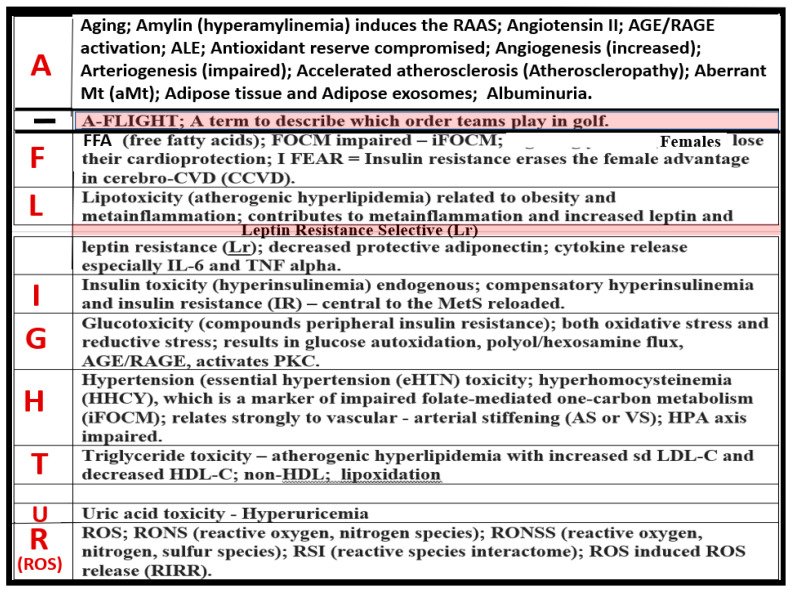
Metabolic syndrome (MetS) is considered a metabolic disorder that has been steadily increasing globally and seems to parallel the increasing prevalence of obesity. It consists of a cluster of risk factors which traditionally includes obesity and hyperlipidemia, hyperinsulinemia, hypertension, and hyperglycemia. These four core risk factors are associated with insulin resistance (IR) and, importantly, the MetS is known to increase the risk for developing cerebrocardiovascular disease and type 2 diabetes mellitus. The MetS had its early origins in IR and syndrome X. It has undergone numerous name changes, with additional risk factors and variables being added over the years; however, it has remained as the MetS worldwide for the past three decades. This overview continues to add novel insights to the MetS and suggests that leptin resistance with hyperleptinemia, aberrant mitochondrial stress and reactive oxygen species (ROS), impaired folate-mediated one-carbon metabolism with hyperhomocysteinemia, vascular stiffening, microalbuminuria, and visceral adipose tissues extracellular vesicle exosomes be added to the list of associated variables. Notably, the role of a dysfunctional and activated endothelium and deficient nitric oxide bioavailability along with a dysfunctional and attenuated endothelial glycocalyx, vascular inflammation, systemic metainflammation, and the important role of ROS and reactive species interactome are discussed. With new insights and knowledge regarding the MetS comes the possibility of new findings through further research.
Keywords: endothelial dysfunction; exosomes; hyperglycemia; hyperinsulinemia; hyperlipidemia; hypertension; insulin resistance; leptin resistance; metainflammation; miRNAs.
Conflict of interest statement
The author declares no conflict of interest.
Figures
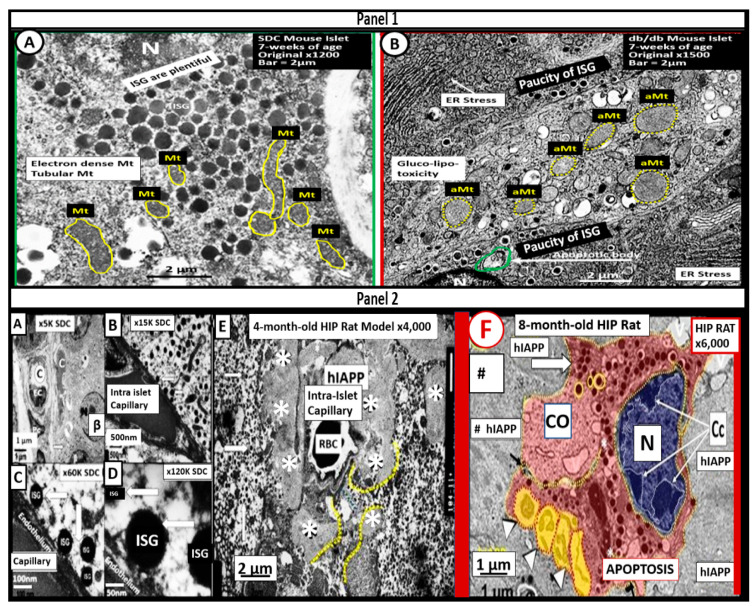
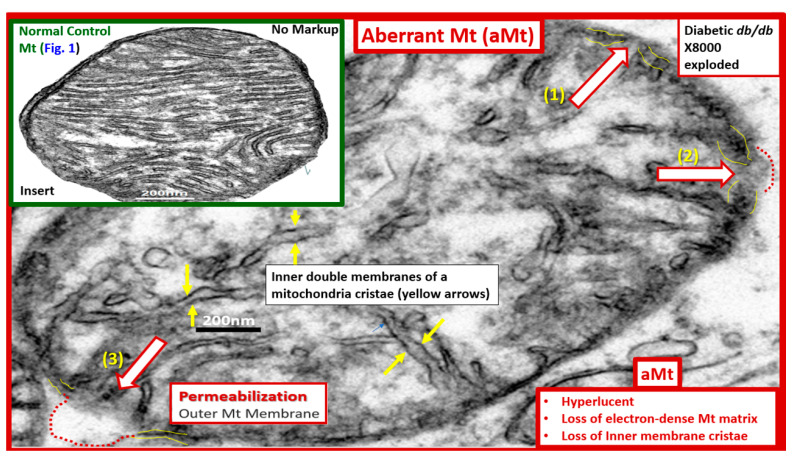
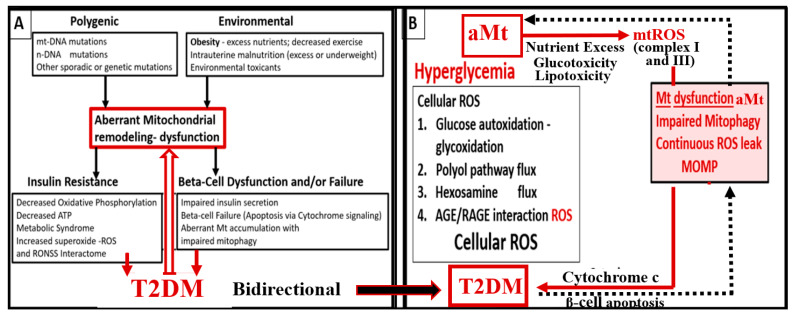
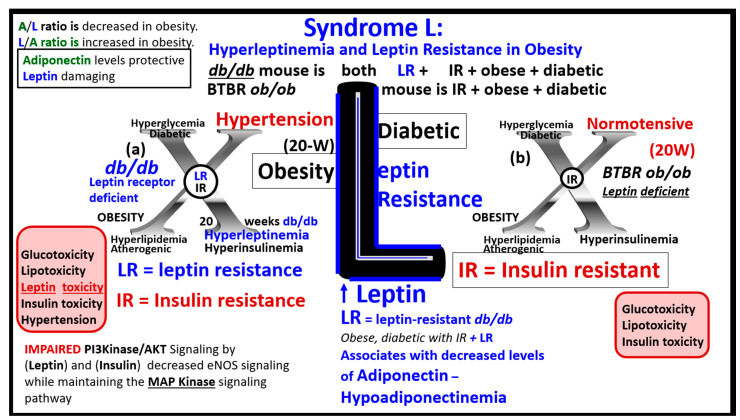



References
-
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults Executive summary of the third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) JAMA. 2002;285:2486–2497. doi: 10.1001/jama.285.19.2486. - DOI - PubMed
-
- Grundy S.M., Brewer H.B., Jr., Cleeman J.I., Smith S.C., Jr., Lenfant C., American Heart Association. National Heart, Lung, and Blood Institute Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. - DOI - PubMed
-
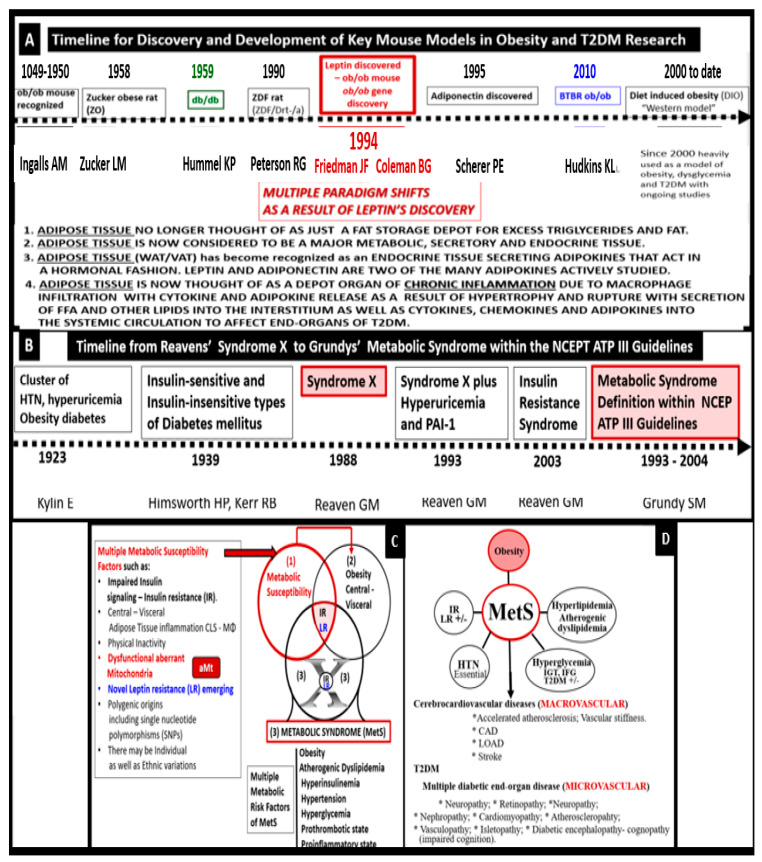
- Kylin E. Studien ueber das Hypertonie-Hyperglyka “mie-Hyperurika” miesyndrom. Zent. Fuer Inn. Med. 1923;44:105–127.
-
- Himsworth H.P., Kerr R.B. Insulin-sensitive and insulin-insensitive types of diabetes mellitus. Clin. Sci. 1939;4:119–152.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
