

Phosphorylation stabilized TET1 acts as an oncoprotein and therapeutic target in B cell acute lymphoblastic leukemia
- PMID: 36989375
- PMCID: PMC11163962
- DOI: 10.1126/scitranslmed.abq8513
Phosphorylation stabilized TET1 acts as an oncoprotein and therapeutic target in B cell acute lymphoblastic leukemia
Abstract
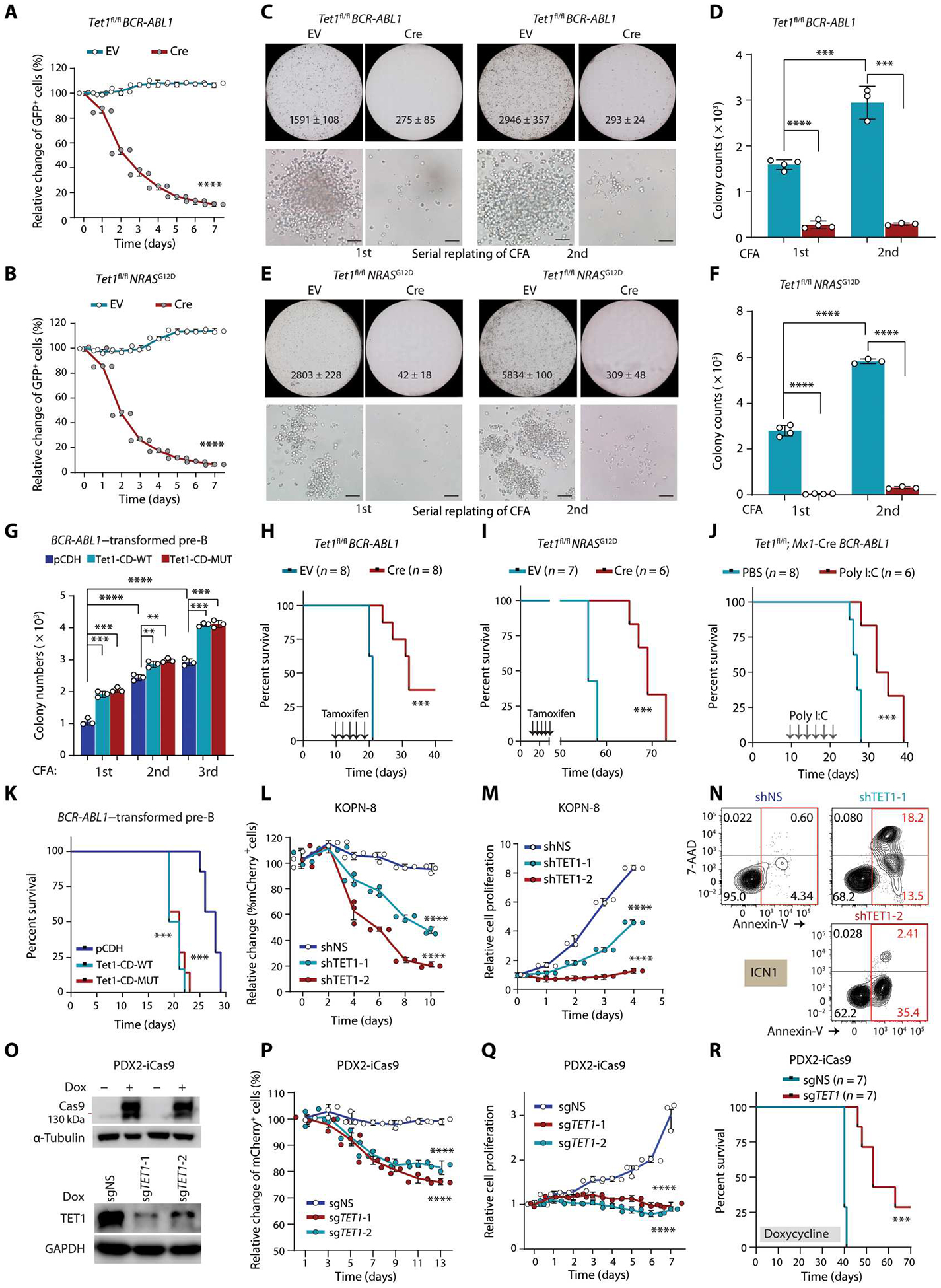
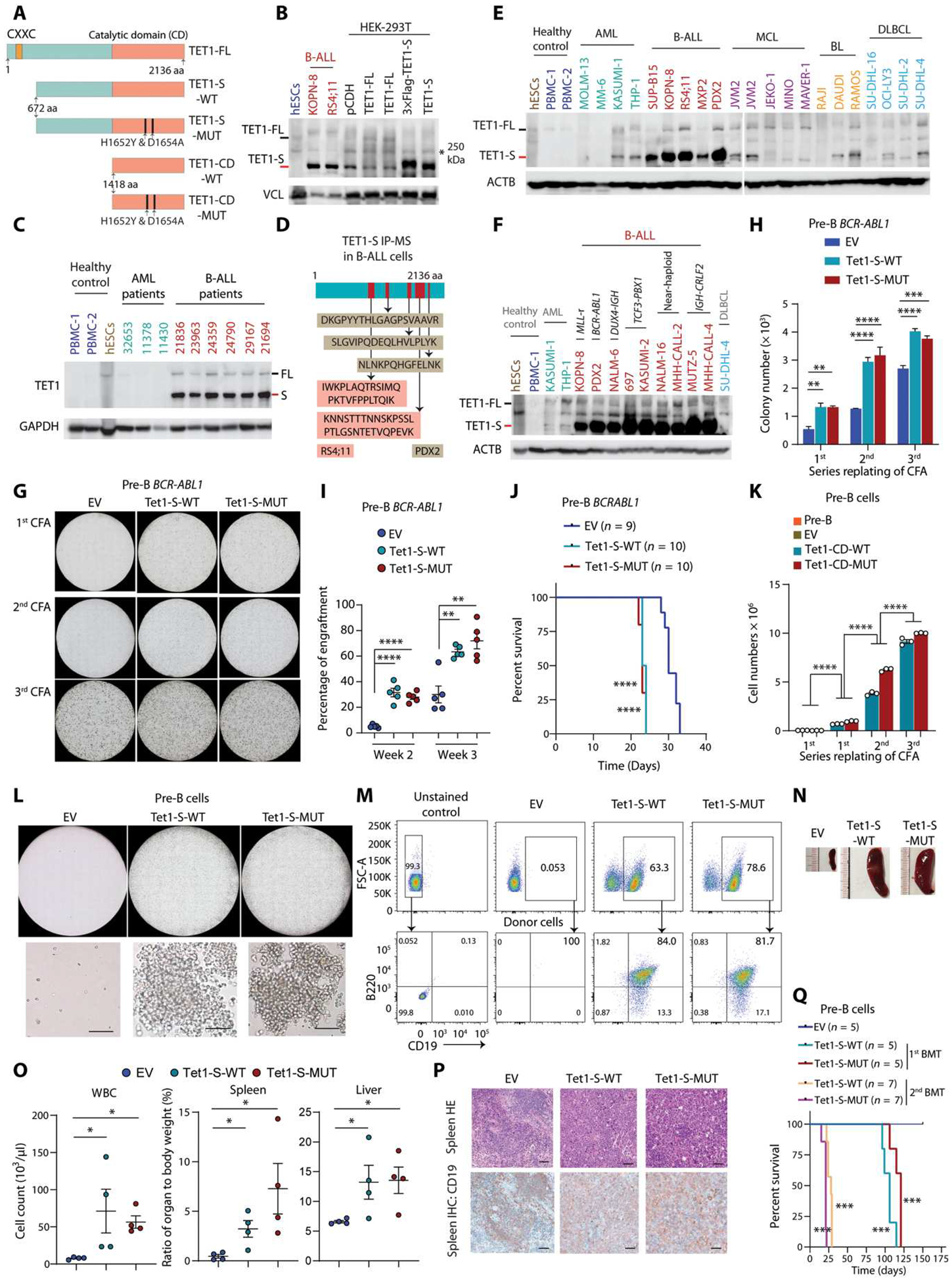
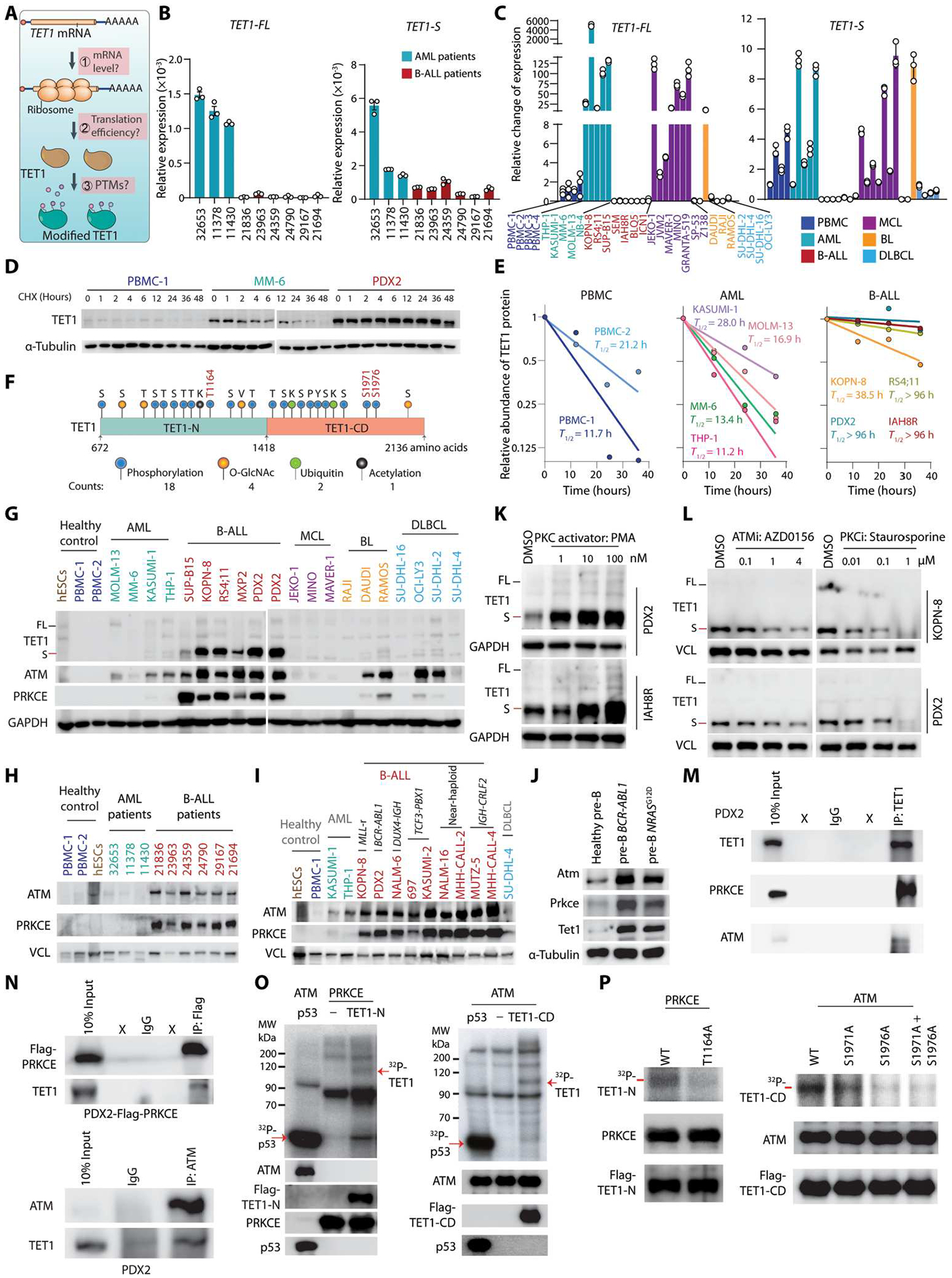
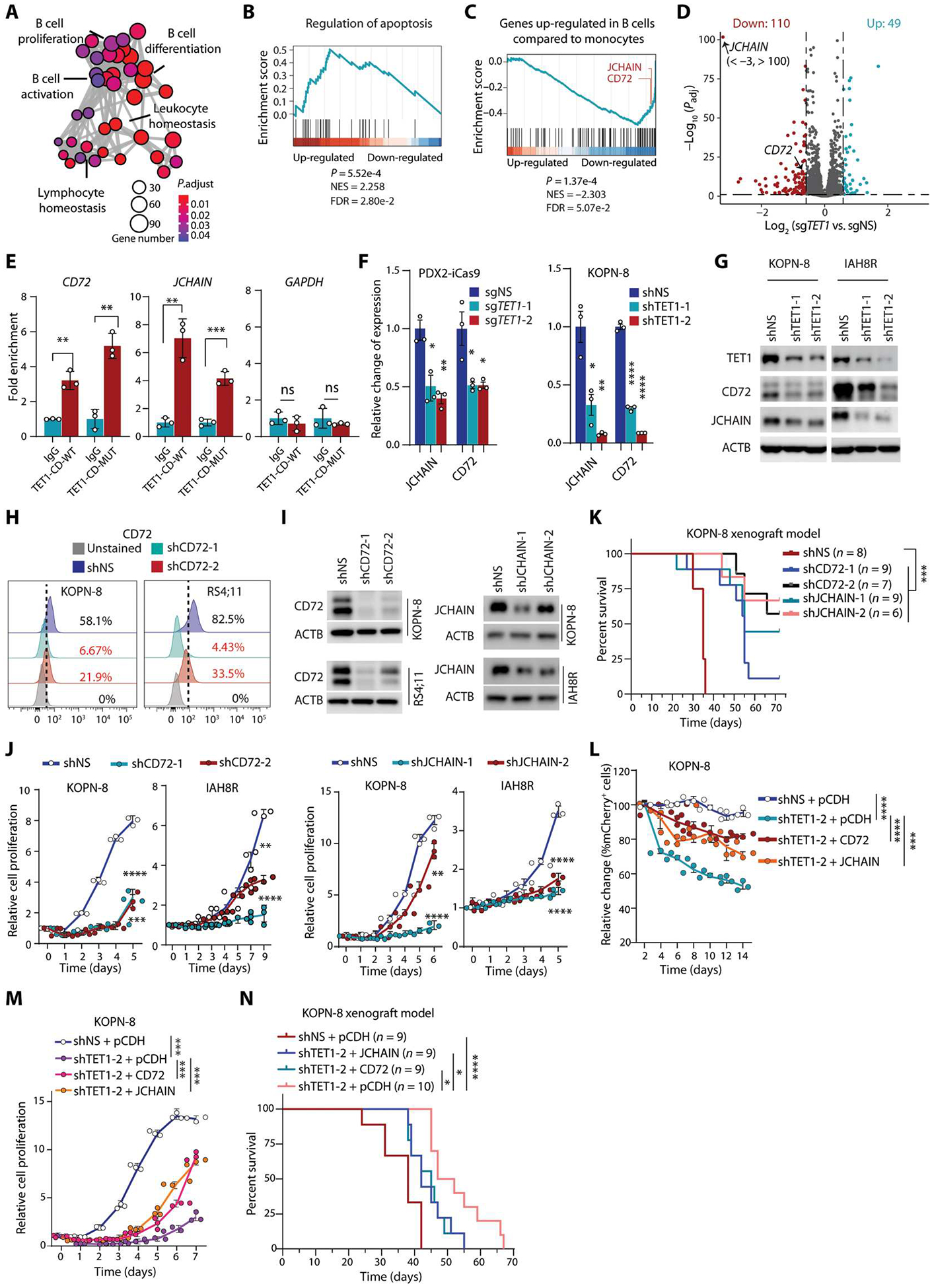
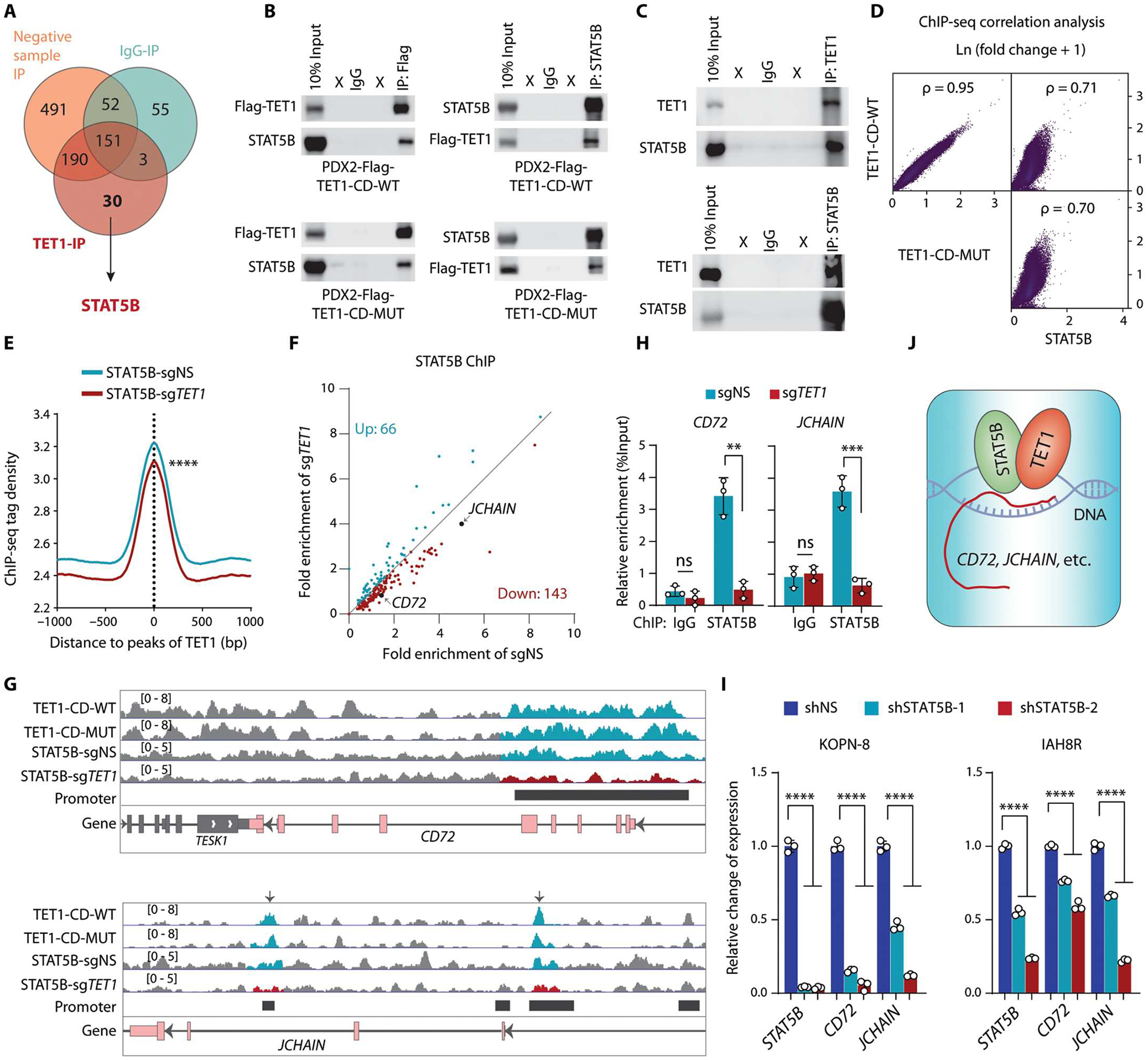
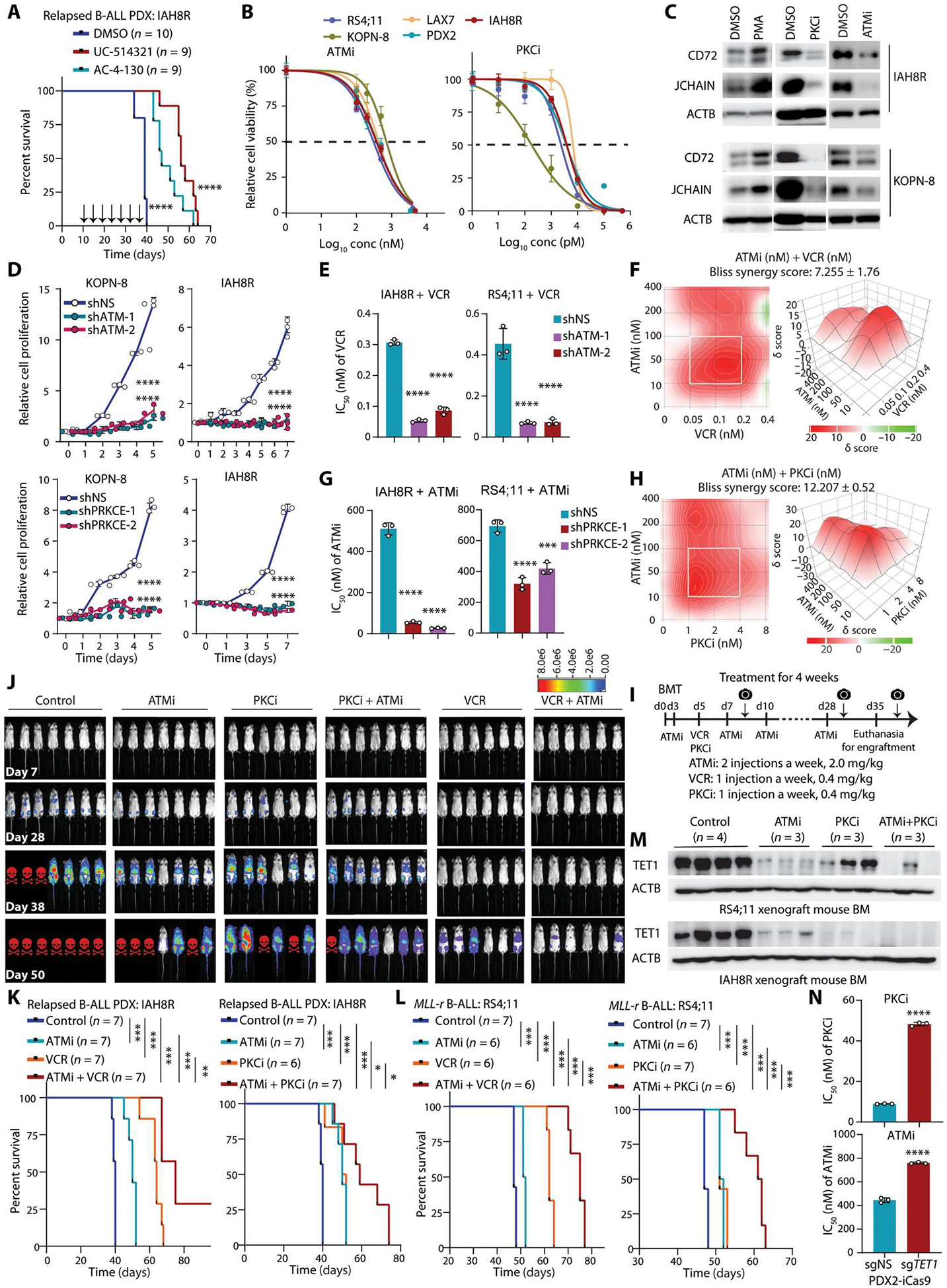
Although the overall survival rate of B cell acute lymphoblastic leukemia (B-ALL) in childhood is more than 80%, it is merely 30% in refractory/relapsed and adult patients with B-ALL. This demonstrates a need for improved therapy targeting this subgroup of B-ALL. Here, we show that the ten-eleven translocation 1 (TET1) protein, a dioxygenase involved in DNA demethylation, is overexpressed and plays a crucial oncogenic role independent of its catalytic activity in B-ALL. Consistent with its oncogenic role in B-ALL, overexpression of TET1 alone in normal precursor B cells is sufficient to transform the cells and cause B-ALL in mice within 3 to 4 months. We found that TET1 protein is stabilized and overexpressed because of its phosphorylation mediated by protein kinase C epsilon (PRKCE) and ATM serine/threonine kinase (ATM), which are also overexpressed in B-ALL. Mechanistically, TET1 recruits STAT5B to the promoters of CD72 and JCHAIN and promotes their transcription, which in turn promotes B-ALL development. Destabilization of TET1 protein by treatment with PKC or ATM inhibitors (staurosporine or AZD0156; both tested in clinical trials), or by pharmacological targeting of STAT5B, greatly decreases B-ALL cell viability and inhibits B-ALL progression in vitro and in vivo. The combination of AZD0156 with staurosporine or vincristine exhibits a synergistic effect on inhibition of refractory/relapsed B-ALL cell survival and leukemia progression in PDX models. Collectively, our study reveals an oncogenic role of the phosphorylated TET1 protein in B-ALL independent of its catalytic activity and highlights the therapeutic potential of targeting TET1 signaling for the treatment of refractory/relapsed B-ALL.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
