

A-MYB and BRDT-dependent RNA Polymerase II pause release orchestrates transcriptional regulation in mammalian meiosis
- PMID: 36990976
- PMCID: PMC10060231
- DOI: 10.1038/s41467-023-37408-w
A-MYB and BRDT-dependent RNA Polymerase II pause release orchestrates transcriptional regulation in mammalian meiosis
Abstract
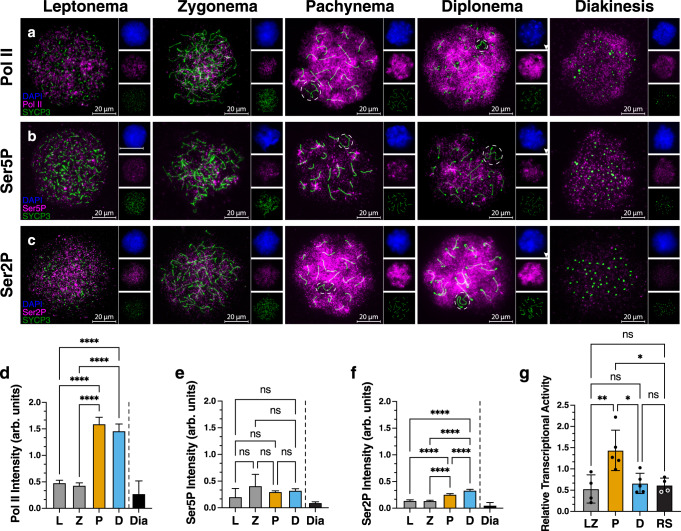
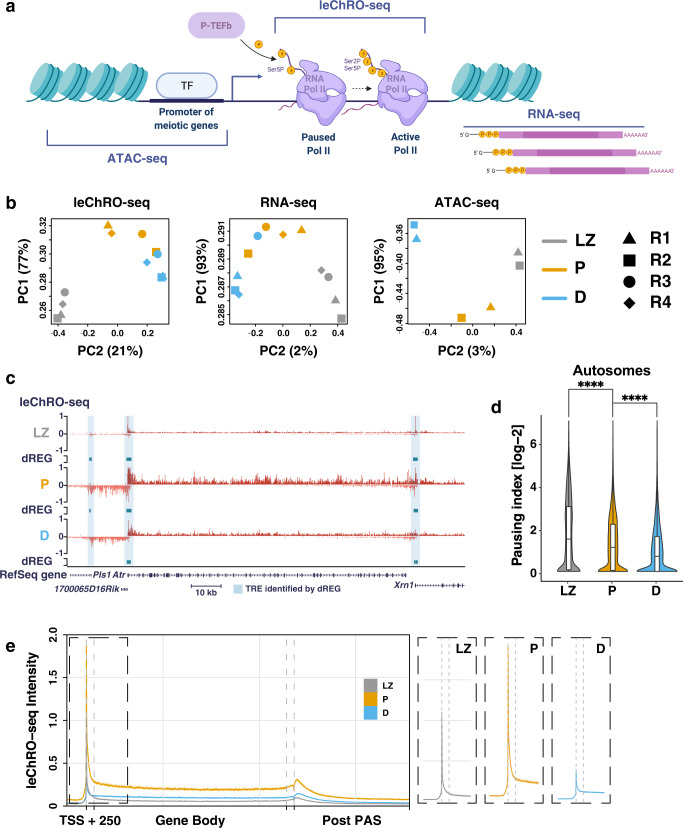
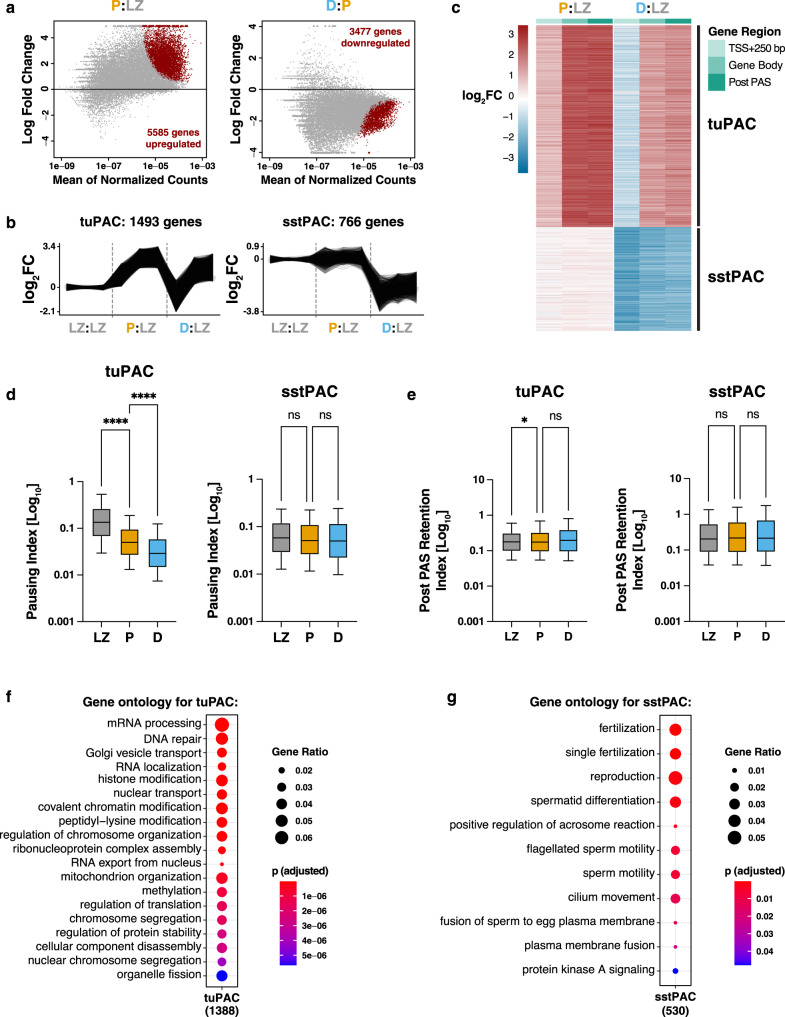
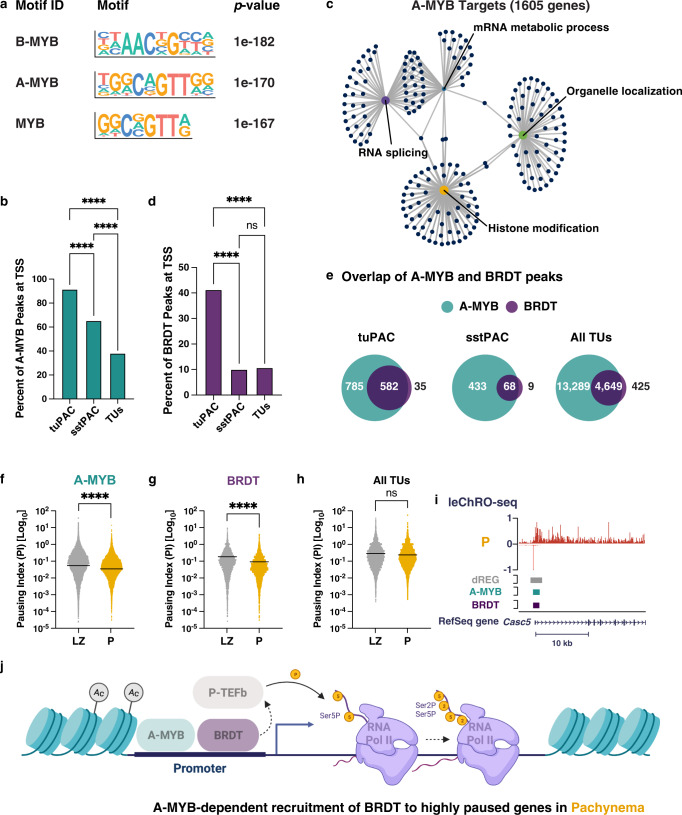
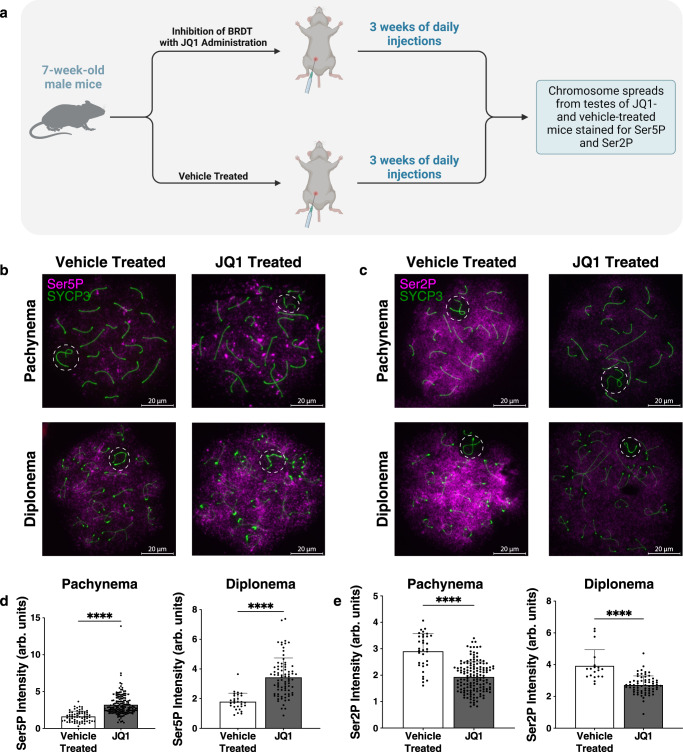
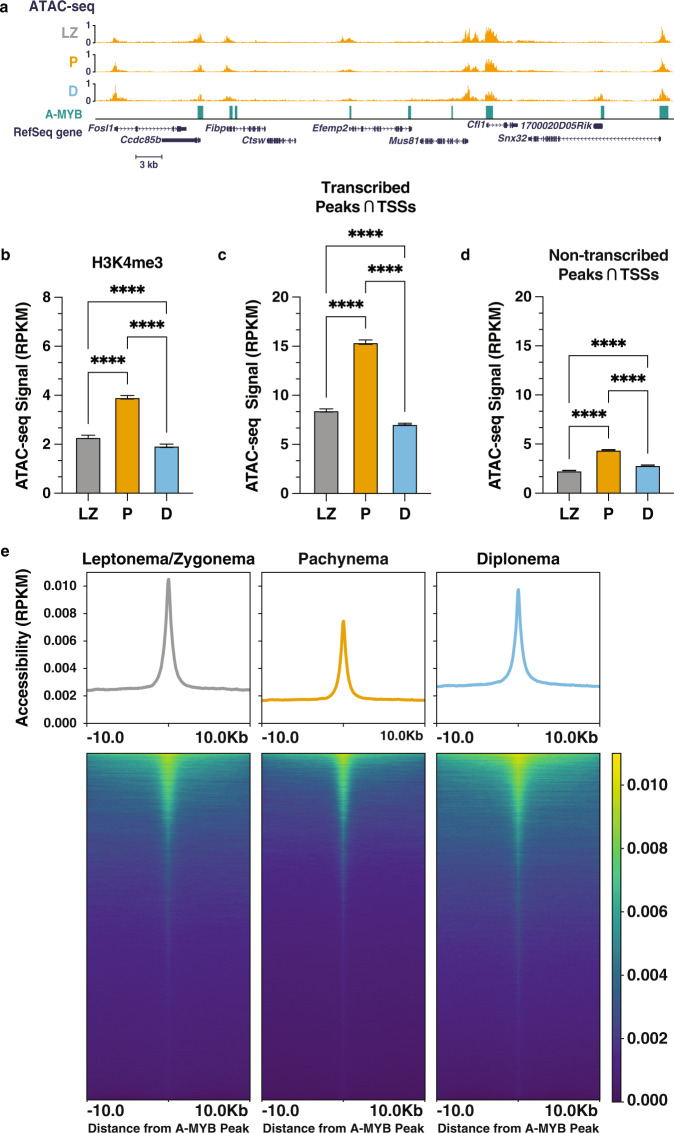
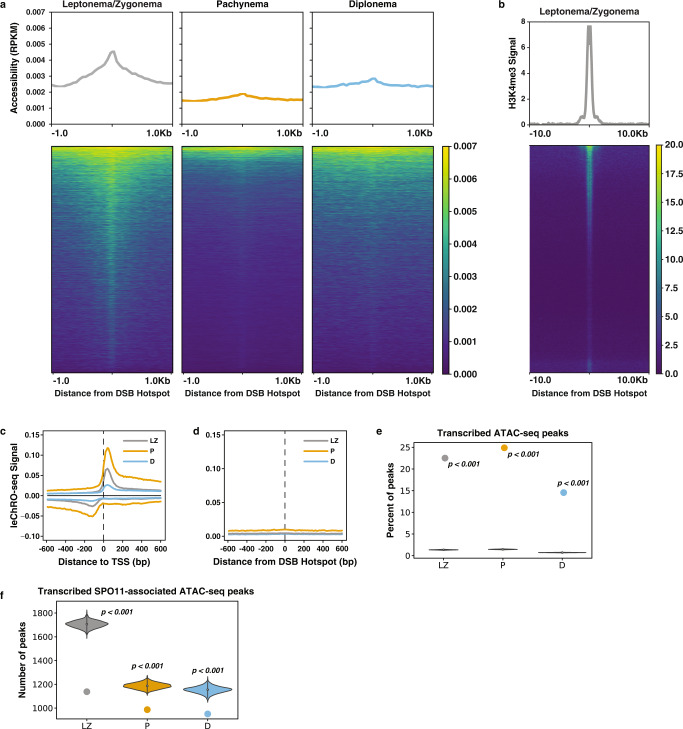
During meiotic prophase I, spermatocytes must balance transcriptional activation with homologous recombination and chromosome synapsis, biological processes requiring extensive changes to chromatin state. We explored the interplay between chromatin accessibility and transcription through prophase I of mammalian meiosis by measuring genome-wide patterns of chromatin accessibility, nascent transcription, and processed mRNA. We find that Pol II is loaded on chromatin and maintained in a paused state early during prophase I. In later stages, paused Pol II is released in a coordinated transcriptional burst mediated by the transcription factors A-MYB and BRDT, resulting in ~3-fold increase in transcription. Transcriptional activity is temporally and spatially segregated from key steps of meiotic recombination: double strand breaks show evidence of chromatin accessibility earlier during prophase I and at distinct loci from those undergoing transcriptional activation, despite shared chromatin marks. Our findings reveal mechanisms underlying chromatin specialization in either transcription or recombination in meiotic cells.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
