

MS2Query: reliable and scalable MS2 mass spectra-based analogue search
- PMID: 36990978
- PMCID: PMC10060387
- DOI: 10.1038/s41467-023-37446-4
MS2Query: reliable and scalable MS2 mass spectra-based analogue search
Abstract
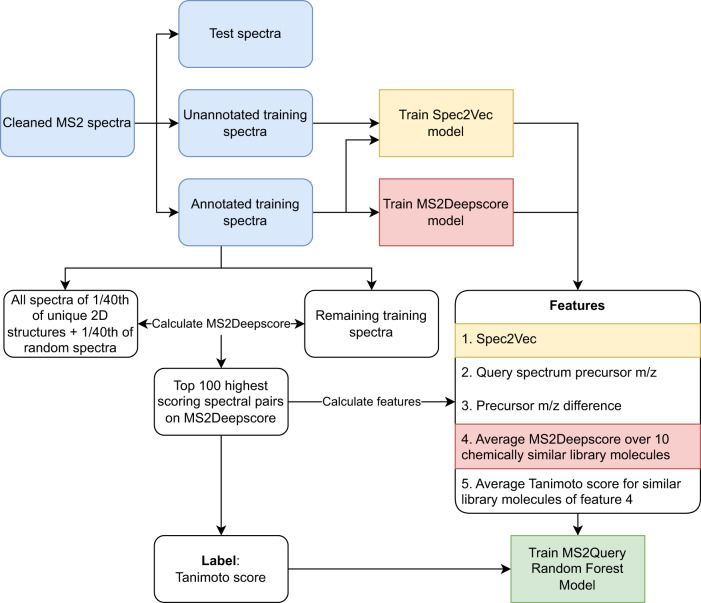
Metabolomics-driven discoveries of biological samples remain hampered by the grand challenge of metabolite annotation and identification. Only few metabolites have an annotated spectrum in spectral libraries; hence, searching only for exact library matches generally returns a few hits. An attractive alternative is searching for so-called analogues as a starting point for structural annotations; analogues are library molecules which are not exact matches but display a high chemical similarity. However, current analogue search implementations are not yet very reliable and relatively slow. Here, we present MS2Query, a machine learning-based tool that integrates mass spectral embedding-based chemical similarity predictors (Spec2Vec and MS2Deepscore) as well as detected precursor masses to rank potential analogues and exact matches. Benchmarking MS2Query on reference mass spectra and experimental case studies demonstrate improved reliability and scalability. Thereby, MS2Query offers exciting opportunities to further increase the annotation rate of metabolomics profiles of complex metabolite mixtures and to discover new biology.
© 2023. The Author(s).
Conflict of interest statement
JJJvdH is currently a member of the Scientific Advisory Board of NAICONS Srl., Milano, Italy. All other authors declare no conflict of interest.
Figures
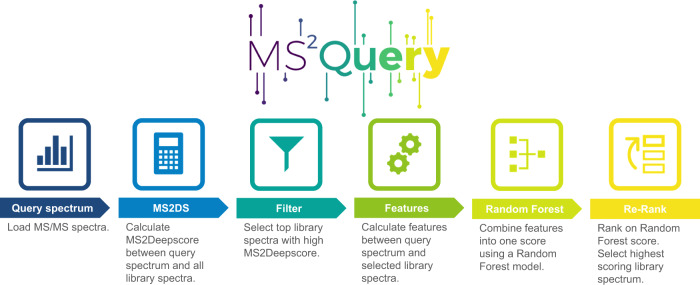
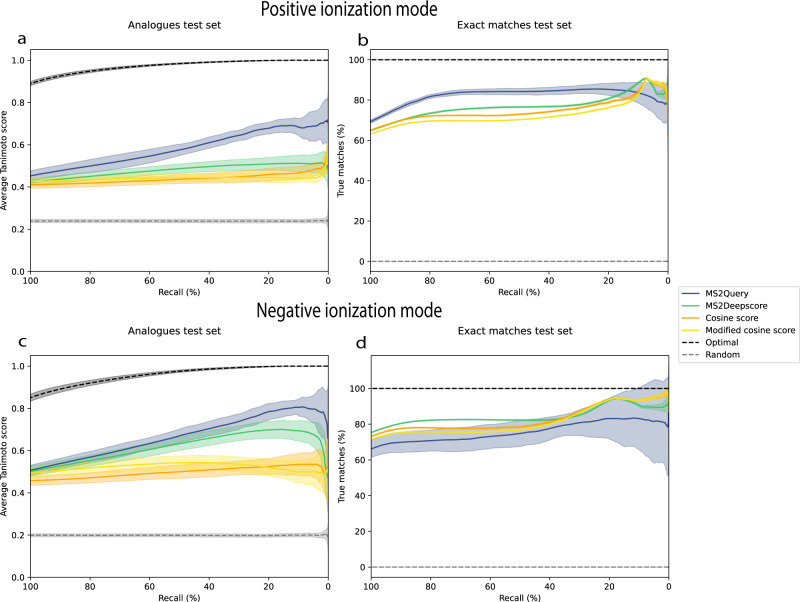
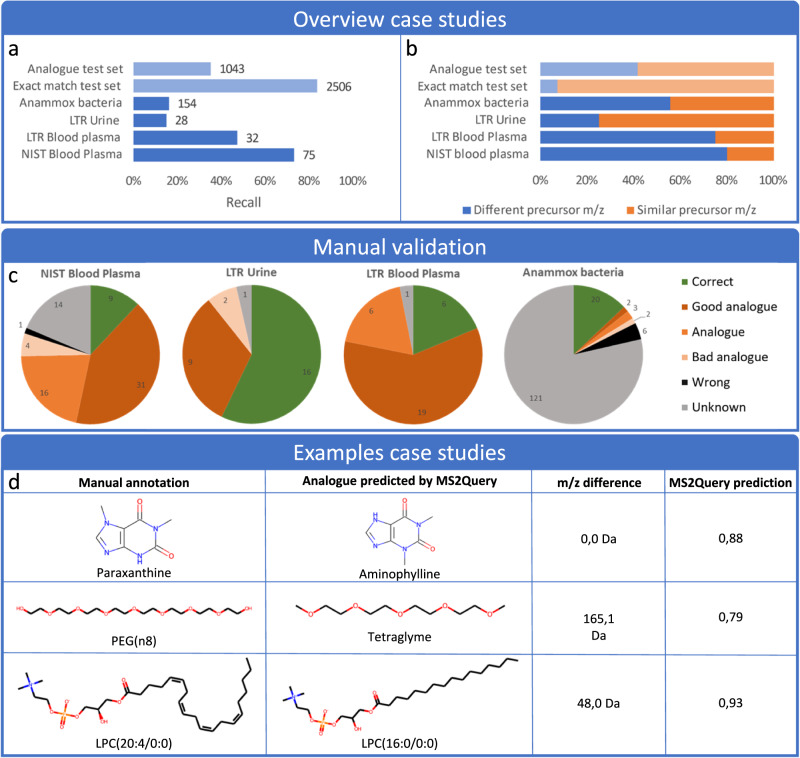
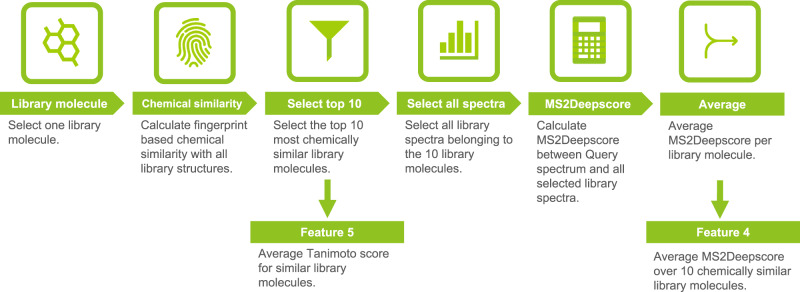
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
