

An atlas of genetic scores to predict multi-omic traits
- PMID: 36991119
- PMCID: PMC10323211
- DOI: 10.1038/s41586-023-05844-9
An atlas of genetic scores to predict multi-omic traits
Abstract
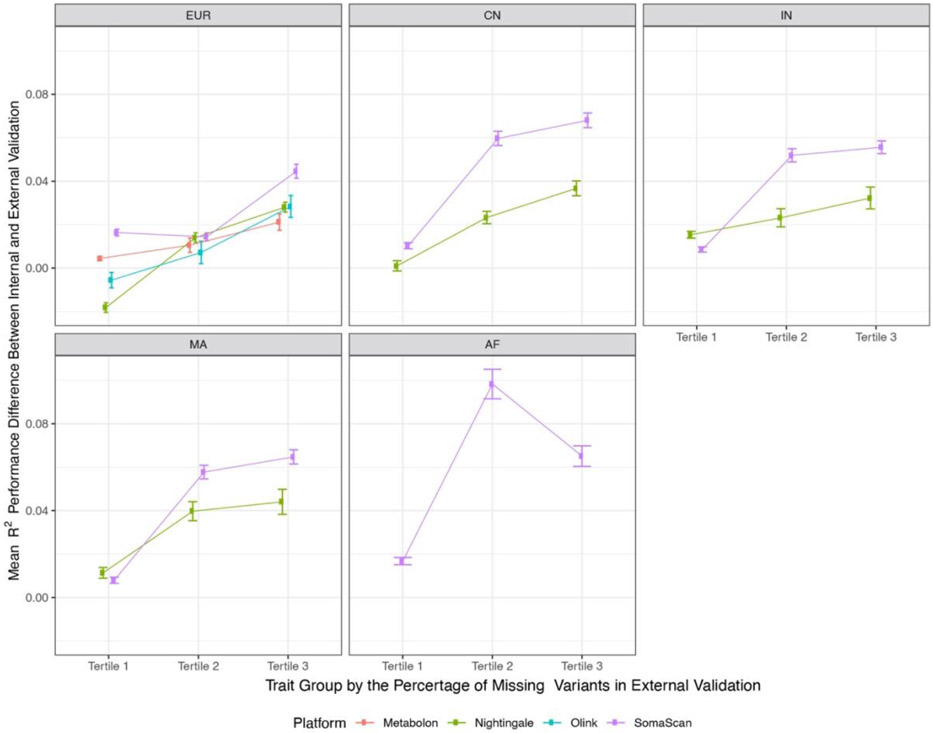
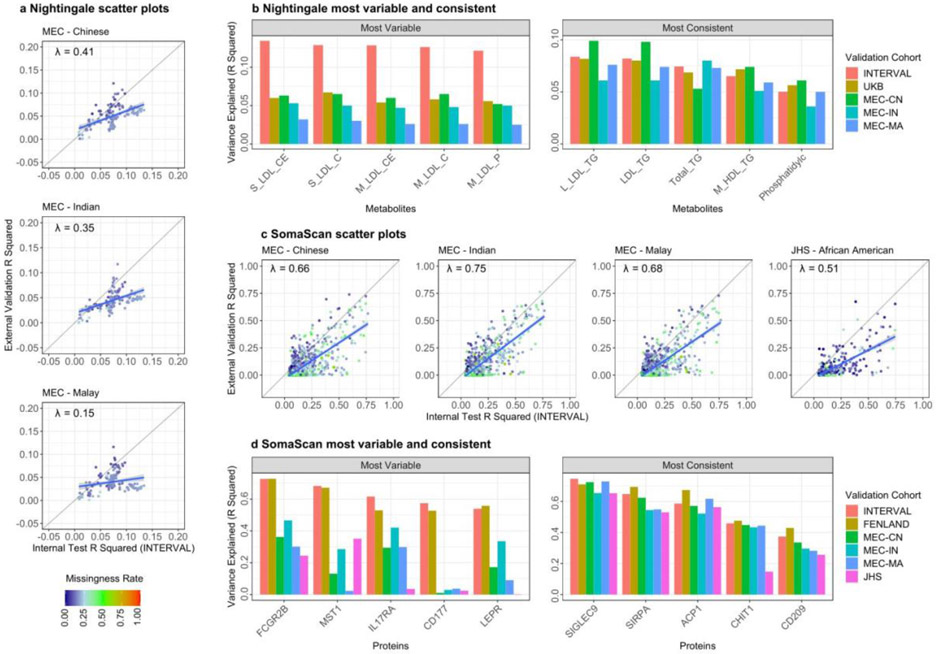
The use of omic modalities to dissect the molecular underpinnings of common diseases and traits is becoming increasingly common. But multi-omic traits can be genetically predicted, which enables highly cost-effective and powerful analyses for studies that do not have multi-omics1. Here we examine a large cohort (the INTERVAL study2; n = 50,000 participants) with extensive multi-omic data for plasma proteomics (SomaScan, n = 3,175; Olink, n = 4,822), plasma metabolomics (Metabolon HD4, n = 8,153), serum metabolomics (Nightingale, n = 37,359) and whole-blood Illumina RNA sequencing (n = 4,136), and use machine learning to train genetic scores for 17,227 molecular traits, including 10,521 that reach Bonferroni-adjusted significance. We evaluate the performance of genetic scores through external validation across cohorts of individuals of European, Asian and African American ancestries. In addition, we show the utility of these multi-omic genetic scores by quantifying the genetic control of biological pathways and by generating a synthetic multi-omic dataset of the UK Biobank3 to identify disease associations using a phenome-wide scan. We highlight a series of biological insights with regard to genetic mechanisms in metabolism and canonical pathway associations with disease; for example, JAK-STAT signalling and coronary atherosclerosis. Finally, we develop a portal ( https://www.omicspred.org/ ) to facilitate public access to all genetic scores and validation results, as well as to serve as a platform for future extensions and enhancements of multi-omic genetic scores.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Figures












Comment in
-
Genetic prediction of multi-omic traits.Nat Rev Genet. 2023 Jun;24(6):346. doi: 10.1038/s41576-023-00602-z. Nat Rev Genet. 2023. PMID: 37016044 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL120393/HL/NHLBI NIH HHS/United States
- HHSN268201800014I/HB/NHLBI NIH HHS/United States
- K08 HL161445/HL/NHLBI NIH HHS/United States
- U01 HL120393/HL/NHLBI NIH HHS/United States
- MC_UU_12015/1/MRC_/Medical Research Council/United Kingdom
- HHSN268201800011C/HL/NHLBI NIH HHS/United States
- HHSN268201000021C/HL/NHLBI NIH HHS/United States
- HHSN268201800015I/HB/NHLBI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 HL117626/HL/NHLBI NIH HHS/United States
- HHSN268201800011I/HB/NHLBI NIH HHS/United States
- MC_UU_00007/10/MRC_/Medical Research Council/United Kingdom
- HHSN268201100037C/HL/NHLBI NIH HHS/United States
- HHSN268201800012I/HB/NHLBI NIH HHS/United States
- HHSN268201800012C/HL/NHLBI NIH HHS/United States
- MR/L003120/1/MRC_/Medical Research Council/United Kingdom
- HHSN268201800014C/HL/NHLBI NIH HHS/United States
- HHSN268201800001C/HL/NHLBI NIH HHS/United States
- MRF-042-0001-RG-PETE-C0839/MRF_/MRF_/United Kingdom
- HHSN268201800013I/MD/NIMHD NIH HHS/United States
- HHSN268201800010I/HB/NHLBI NIH HHS/United States
- MC_UU_00006/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
