

The shaky foundations of simulating single-cell RNA sequencing data
- PMID: 36991470
- PMCID: PMC10061781
- DOI: 10.1186/s13059-023-02904-1
The shaky foundations of simulating single-cell RNA sequencing data
Erratum in
-
Author Correction: The shaky foundations of simulating single-cell RNA sequencing data.Genome Biol. 2024 Jul 5;25(1):178. doi: 10.1186/s13059-024-03329-0. Genome Biol. 2024. PMID: 38970115 Free PMC article. No abstract available.
Abstract
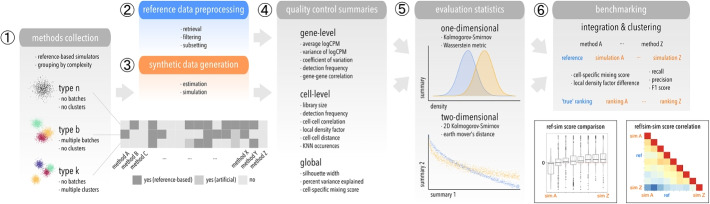
Background: With the emergence of hundreds of single-cell RNA-sequencing (scRNA-seq) datasets, the number of computational tools to analyze aspects of the generated data has grown rapidly. As a result, there is a recurring need to demonstrate whether newly developed methods are truly performant-on their own as well as in comparison to existing tools. Benchmark studies aim to consolidate the space of available methods for a given task and often use simulated data that provide a ground truth for evaluations, thus demanding a high quality standard results credible and transferable to real data.
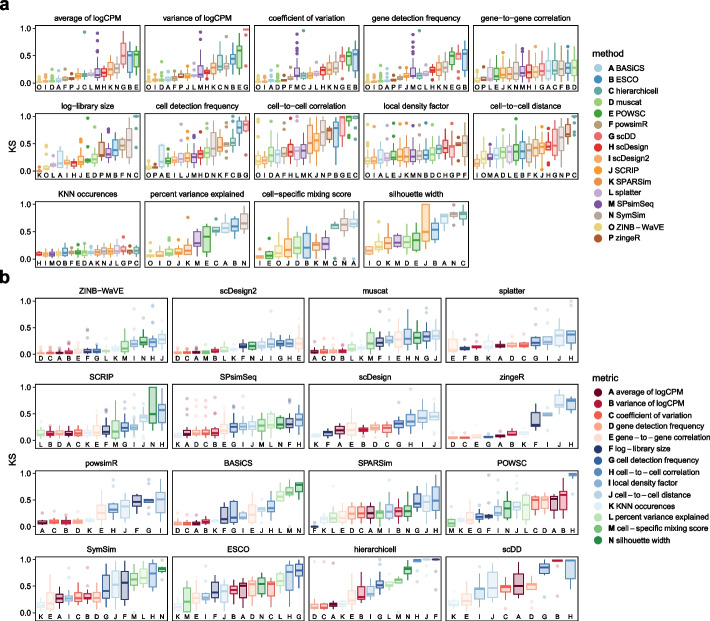
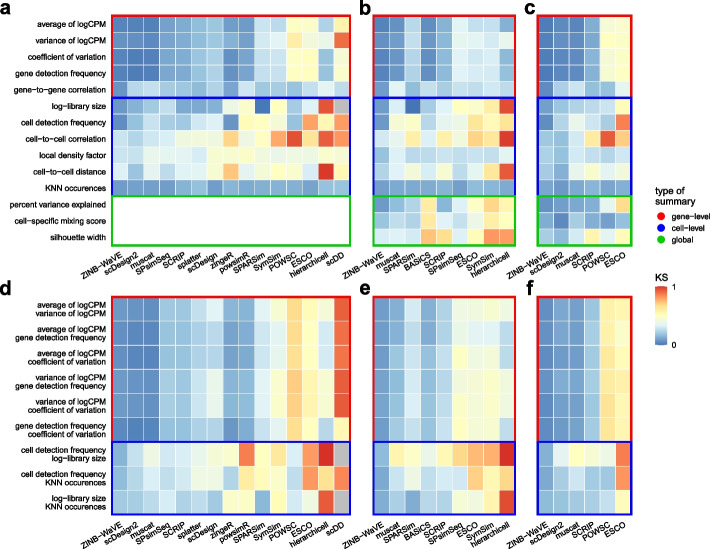
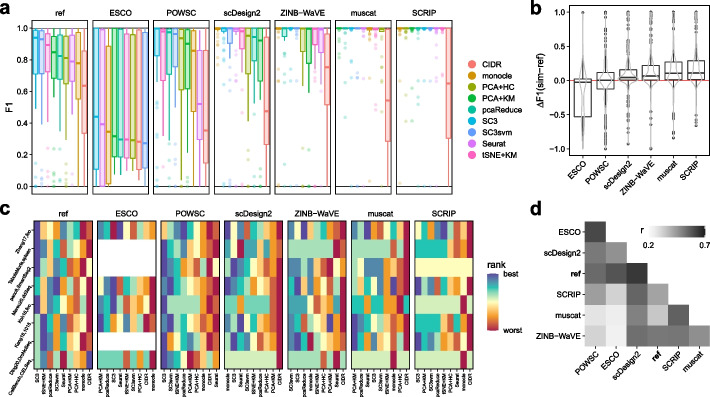
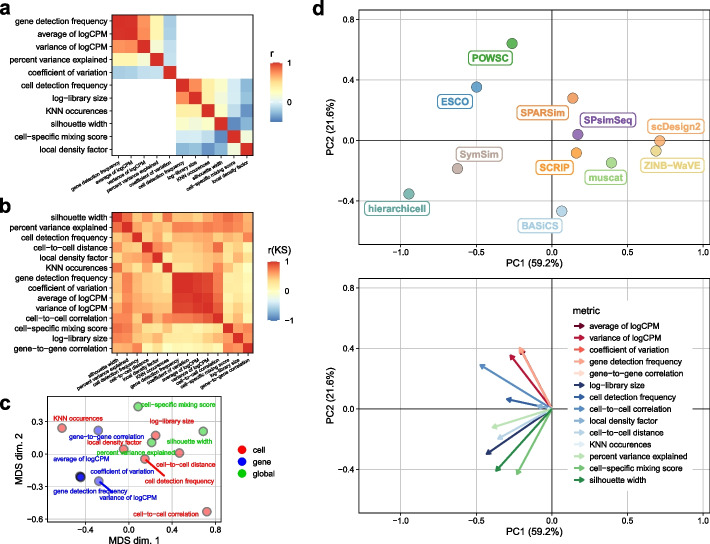
Results: Here, we evaluated methods for synthetic scRNA-seq data generation in their ability to mimic experimental data. Besides comparing gene- and cell-level quality control summaries in both one- and two-dimensional settings, we further quantified these at the batch- and cluster-level. Secondly, we investigate the effect of simulators on clustering and batch correction method comparisons, and, thirdly, which and to what extent quality control summaries can capture reference-simulation similarity.
Conclusions: Our results suggest that most simulators are unable to accommodate complex designs without introducing artificial effects, they yield over-optimistic performance of integration and potentially unreliable ranking of clustering methods, and it is generally unknown which summaries are important to ensure effective simulation-based method comparisons.
Keywords: Benchmarking; Simulation; Single-cell RNA-seq.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Comment in
-
Simulating scRNA-seq for benchmarks.Nat Genet. 2023 Jun;55(6):904. doi: 10.1038/s41588-023-01431-w. Nat Genet. 2023. PMID: 37308673 No abstract available.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
