

This is a preprint.
Mapping the dynamic genetic regulatory architecture of HLA genes at single-cell resolution
- PMID: 36993194
- PMCID: PMC10055604
- DOI: 10.1101/2023.03.14.23287257
Mapping the dynamic genetic regulatory architecture of HLA genes at single-cell resolution
Update in
-
Mapping the dynamic genetic regulatory architecture of HLA genes at single-cell resolution.Nat Genet. 2023 Dec;55(12):2255-2268. doi: 10.1038/s41588-023-01586-6. Epub 2023 Nov 30. Nat Genet. 2023. PMID: 38036787 Free PMC article.
Abstract
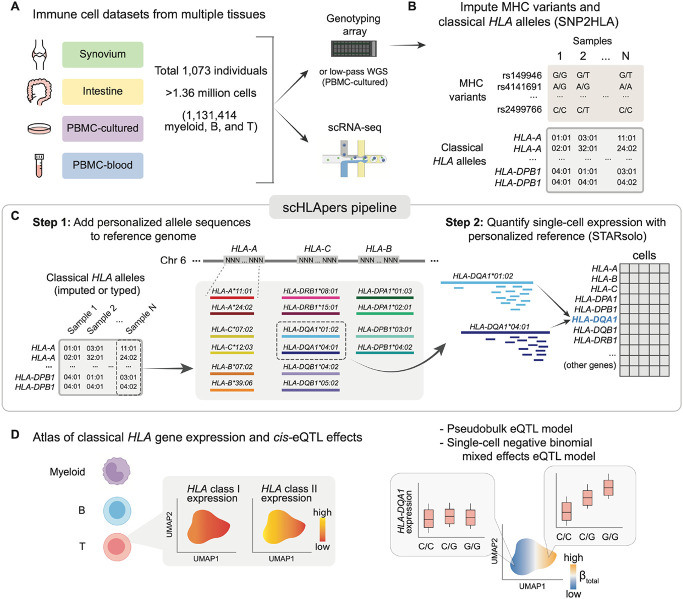
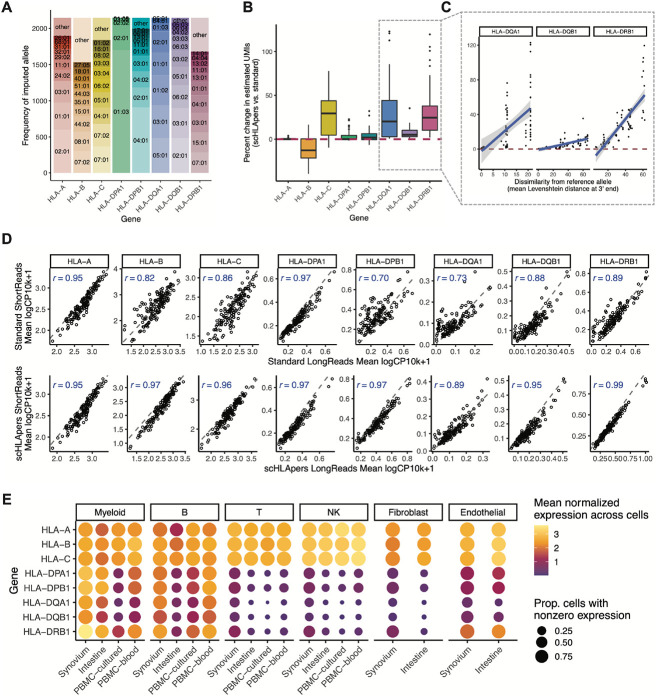
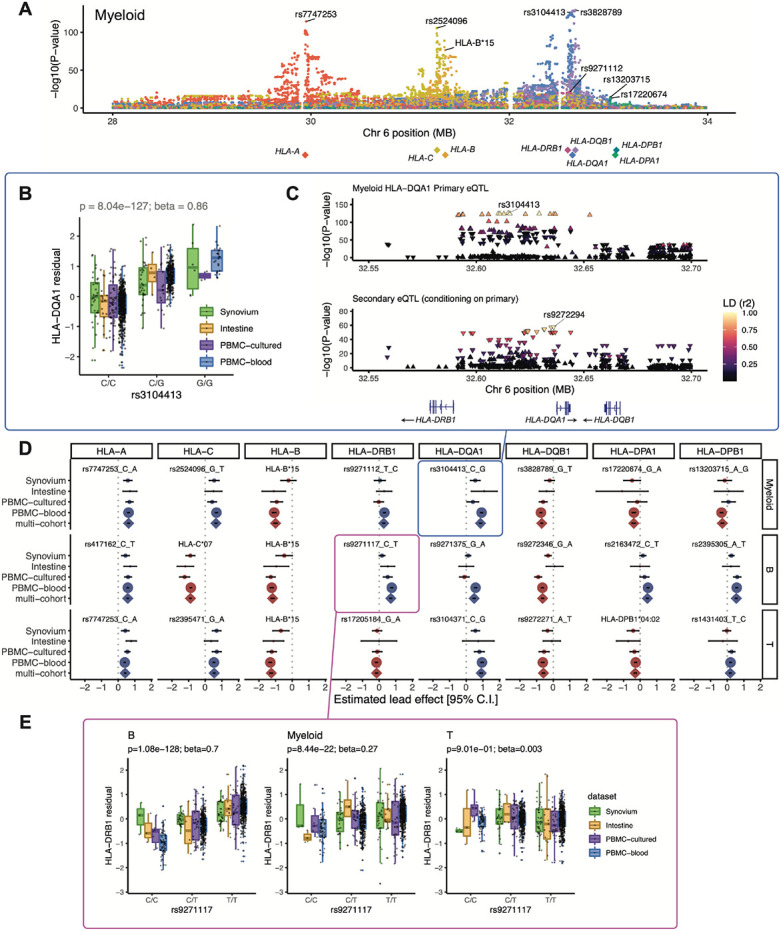
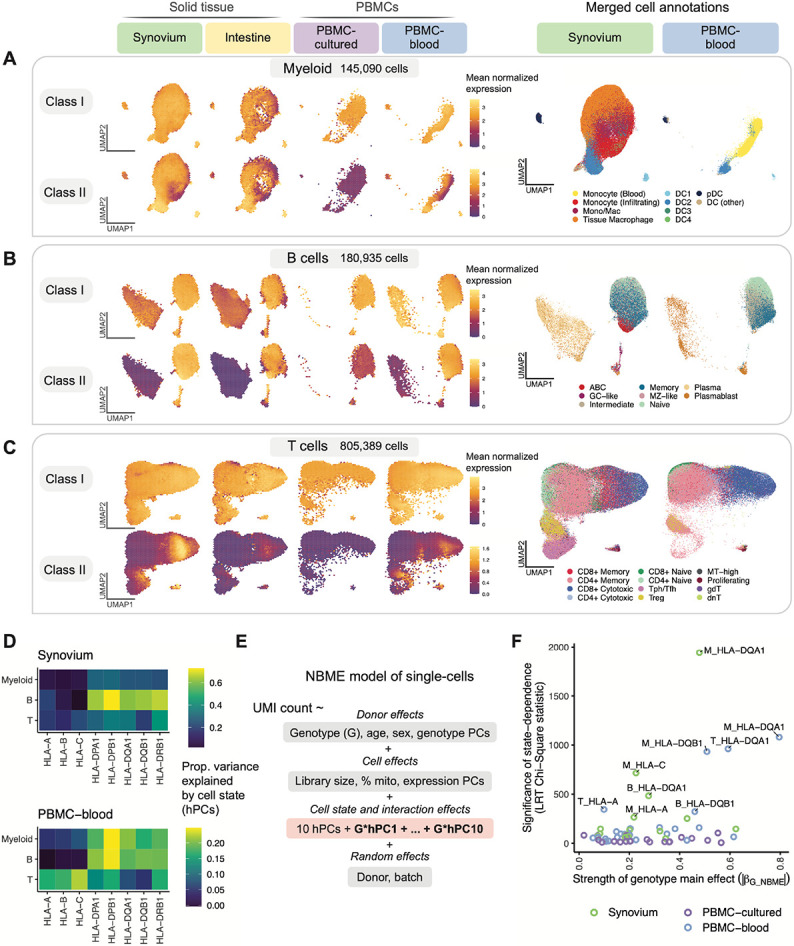
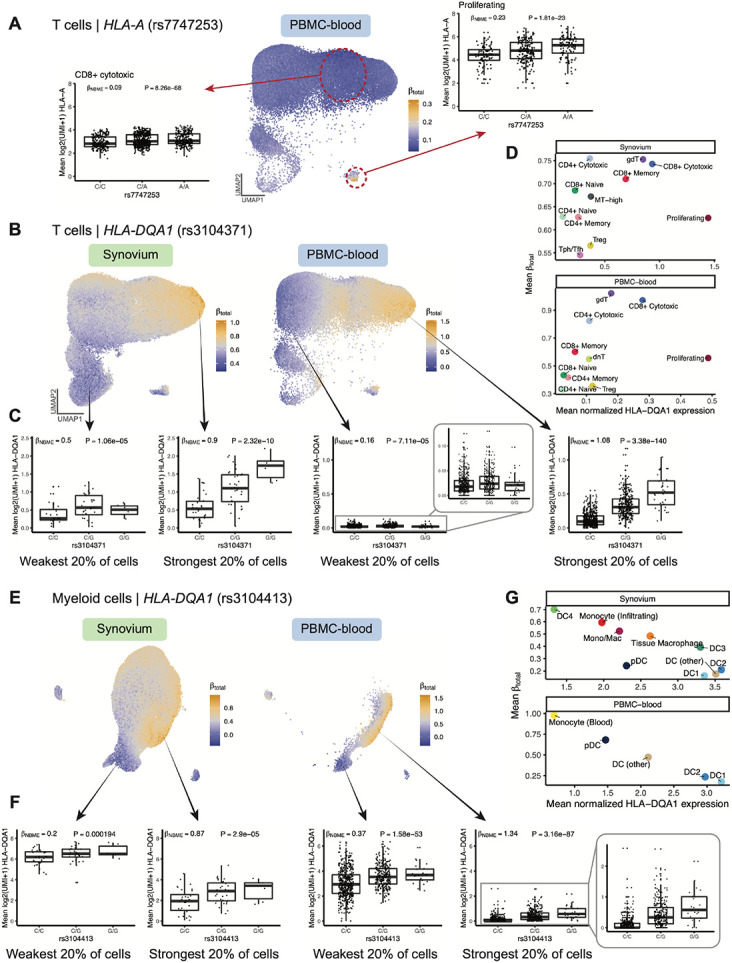
The human leukocyte antigen (HLA) locus plays a critical role in complex traits spanning autoimmune and infectious diseases, transplantation, and cancer. While coding variation in HLA genes has been extensively documented, regulatory genetic variation modulating HLA expression levels has not been comprehensively investigated. Here, we mapped expression quantitative trait loci (eQTLs) for classical HLA genes across 1,073 individuals and 1,131,414 single cells from three tissues, using personalized reference genomes to mitigate technical confounding. We identified cell-type-specific cis-eQTLs for every classical HLA gene. Modeling eQTLs at single-cell resolution revealed that many eQTL effects are dynamic across cell states even within a cell type. HLA-DQ genes exhibit particularly cell-state-dependent effects within myeloid, B, and T cells. Dynamic HLA regulation may underlie important interindividual variability in immune responses.
Figures
References
-
- Dendrou C. A., Petersen J., Rossjohn J., Fugger L., HLA variation and disease. Nat. Rev. Immunol. 18, 325–339 (2018). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials