

This is a preprint.
BRCA1 secondary splice-site mutations drive exon-skipping and PARP inhibitor resistance
- PMID: 36993400
- PMCID: PMC10055590
- DOI: 10.1101/2023.03.20.23287465
BRCA1 secondary splice-site mutations drive exon-skipping and PARP inhibitor resistance
Update in
-
BRCA1 secondary splice-site mutations drive exon-skipping and PARP inhibitor resistance.Mol Cancer. 2024 Aug 5;23(1):158. doi: 10.1186/s12943-024-02048-1. Mol Cancer. 2024. PMID: 39103848 Free PMC article.
Abstract
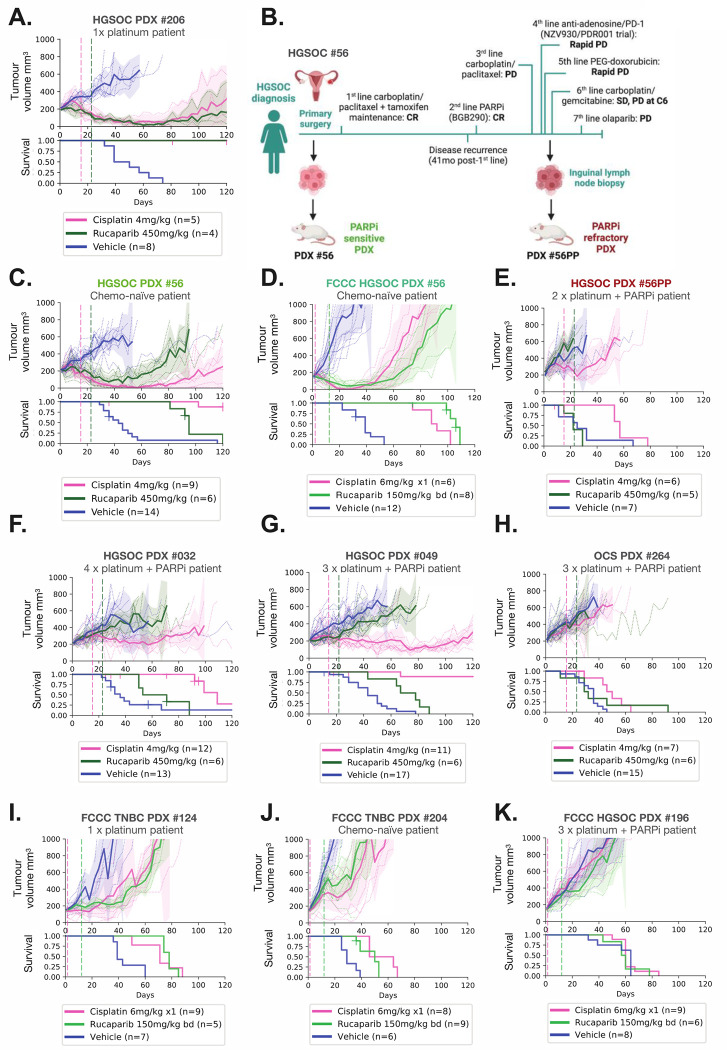
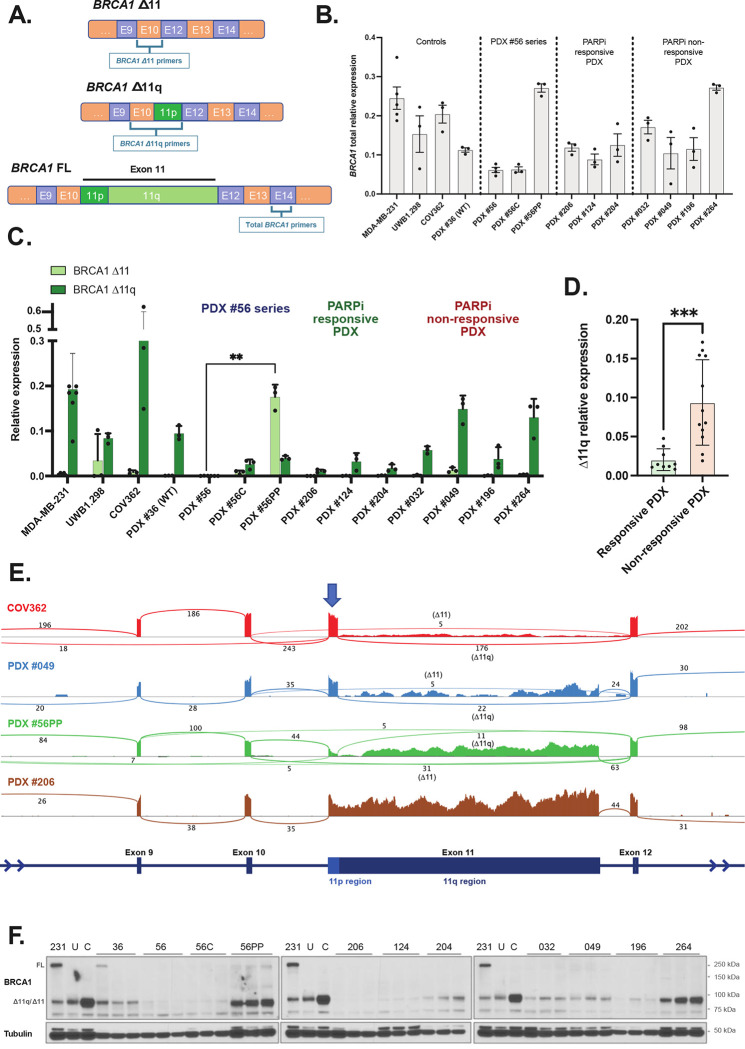
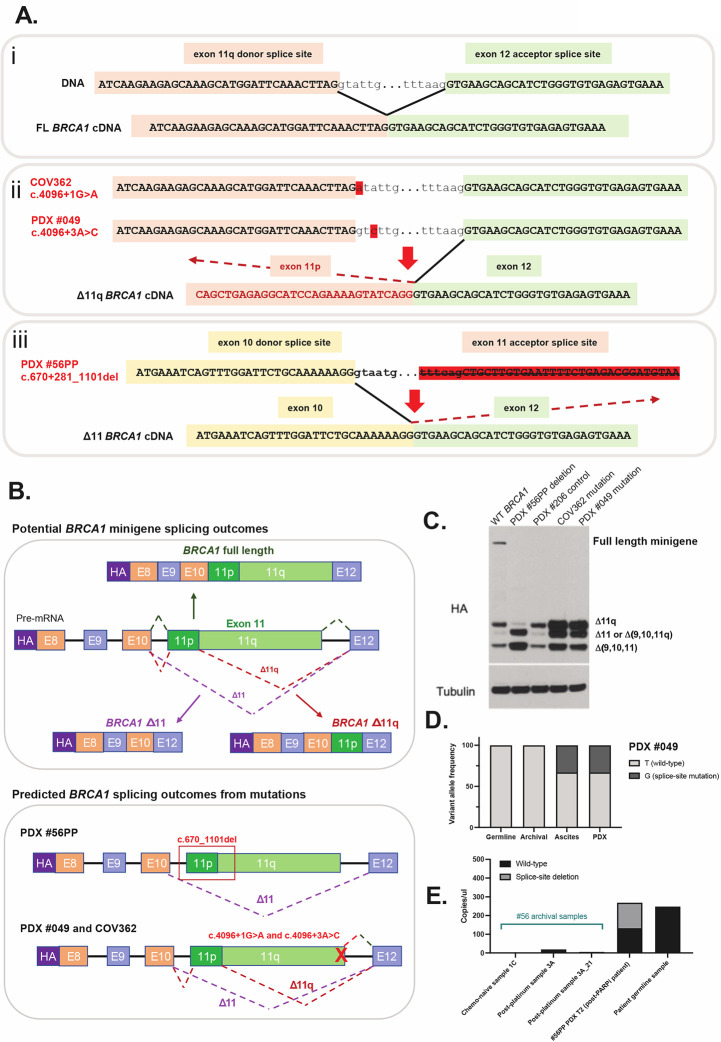
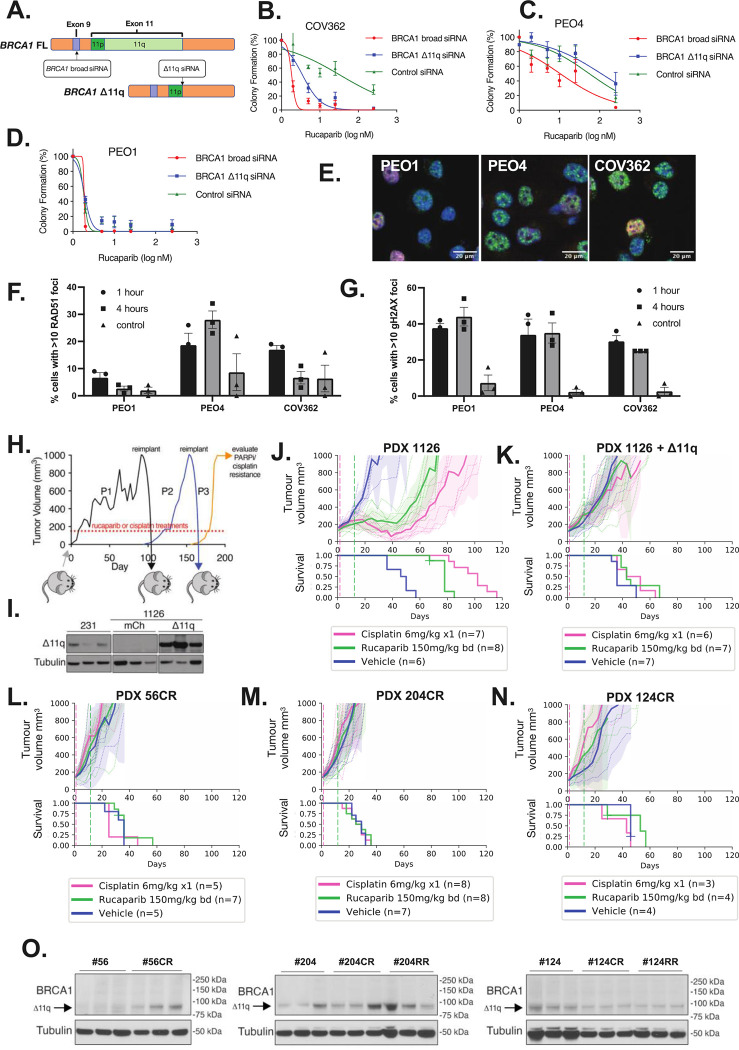
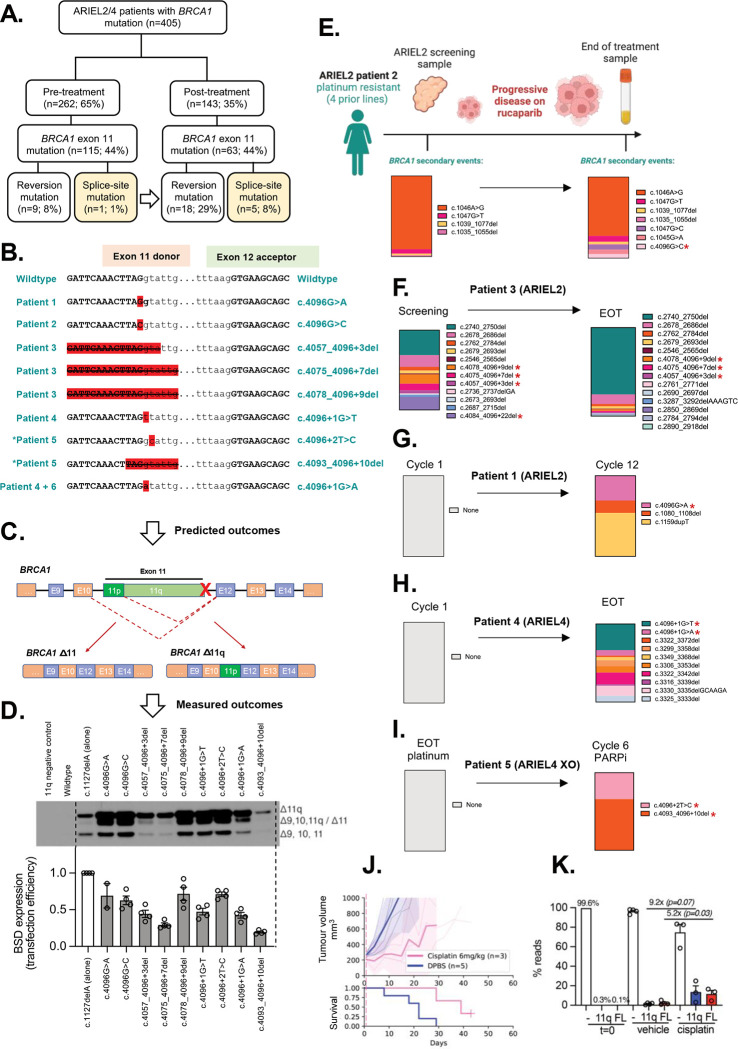
BRCA1 splice isoforms Δ11 and Δ11q can contribute to PARP inhibitor (PARPi) resistance by splicing-out the mutation-containing exon, producing truncated, partially-functional proteins. However, the clinical impact and underlying drivers of BRCA1 exon skipping remain undetermined. We analyzed nine ovarian and breast cancer patient derived xenografts (PDX) with BRCA1 exon 11 frameshift mutations for exon skipping and therapy response, including a matched PDX pair derived from a patient pre- and post-chemotherapy/PARPi. BRCA1 exon 11 skipping was elevated in PARPi resistant PDX tumors. Two independent PDX models acquired secondary BRCA1 splice site mutations (SSMs), predicted in silico to drive exon skipping. Predictions were confirmed using qRT-PCR, RNA sequencing, western blots and BRCA1 minigene modelling. SSMs were also enriched in post-PARPi ovarian cancer patient cohorts from the ARIEL2 and ARIEL4 clinical trials. We demonstrate that SSMs drive BRCA1 exon 11 skipping and PARPi resistance, and should be clinically monitored, along with frame-restoring secondary mutations.
Figures
References
-
- Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. New England Journal of Medicine 2019;381(25):2416–28. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous