

This is a preprint.
Multiomics integration of 22 immune-mediated monogenic diseases reveals an emergent axis of human immune health
- PMID: 36993430
- PMCID: PMC10055521
- DOI: 10.21203/rs.3.rs-2070975/v1
Multiomics integration of 22 immune-mediated monogenic diseases reveals an emergent axis of human immune health
Update in
-
A unified metric of human immune health.Nat Med. 2024 Sep;30(9):2461-2472. doi: 10.1038/s41591-024-03092-6. Epub 2024 Jul 3. Nat Med. 2024. PMID: 38961223 Free PMC article.
Abstract
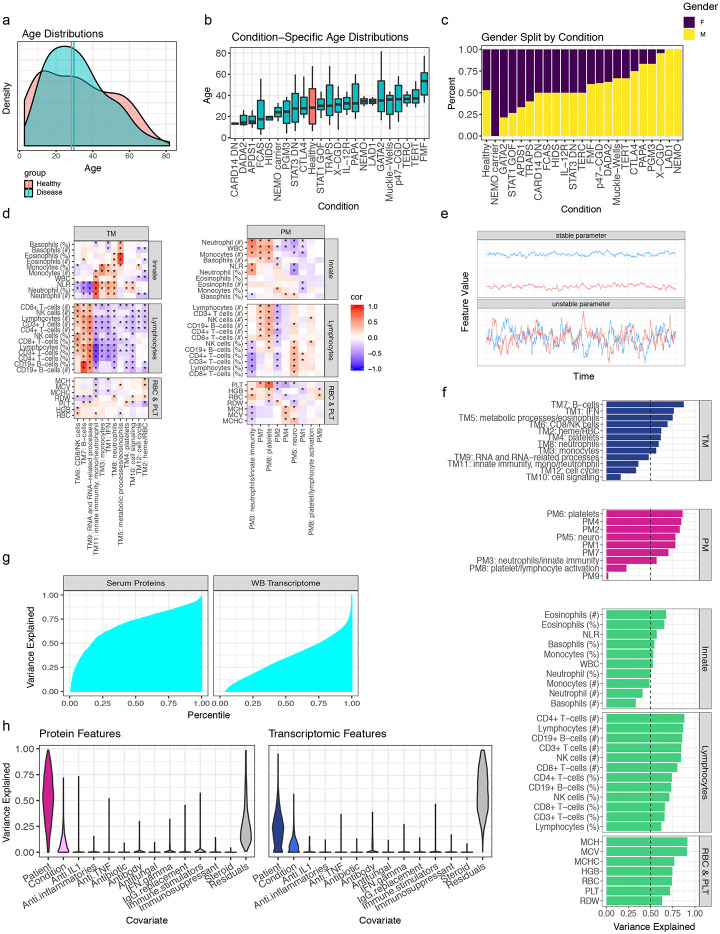
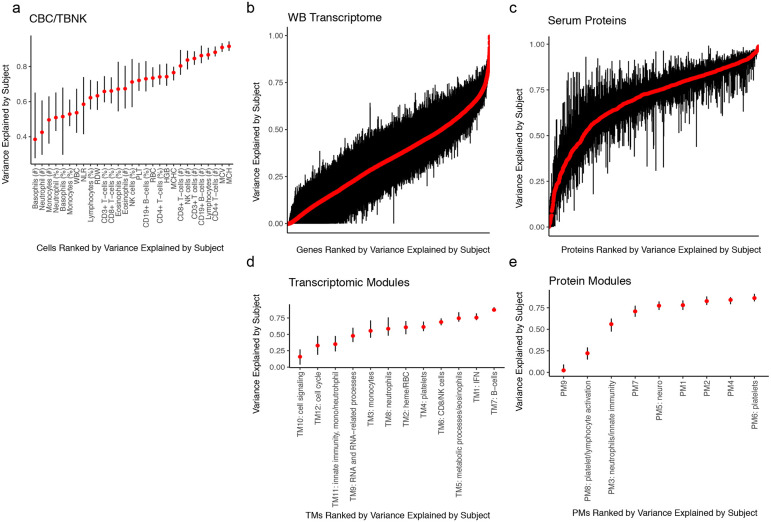
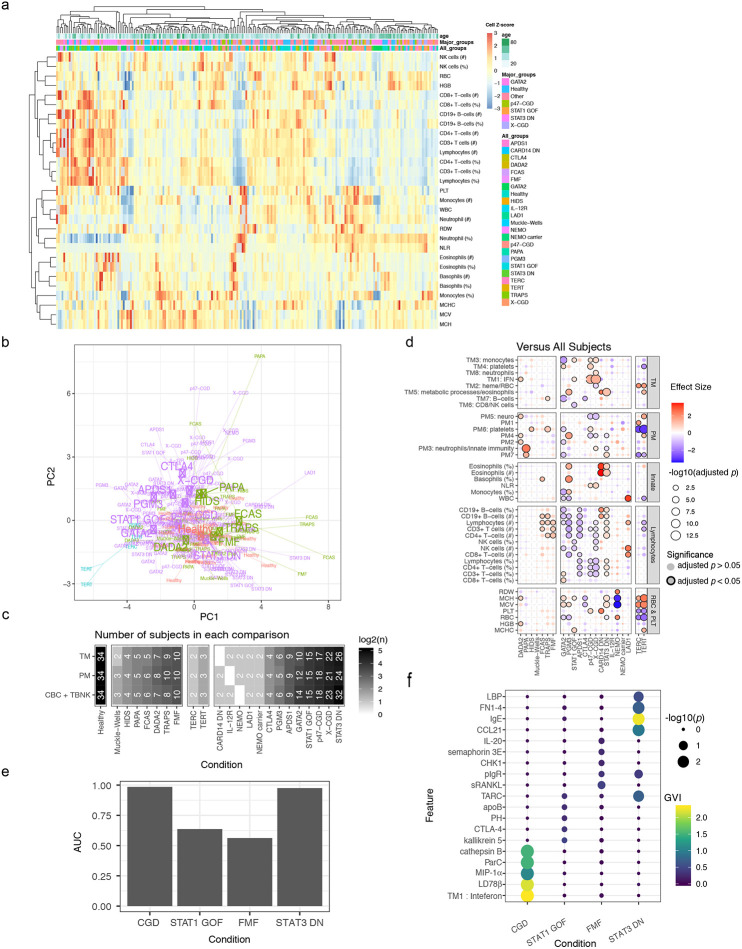
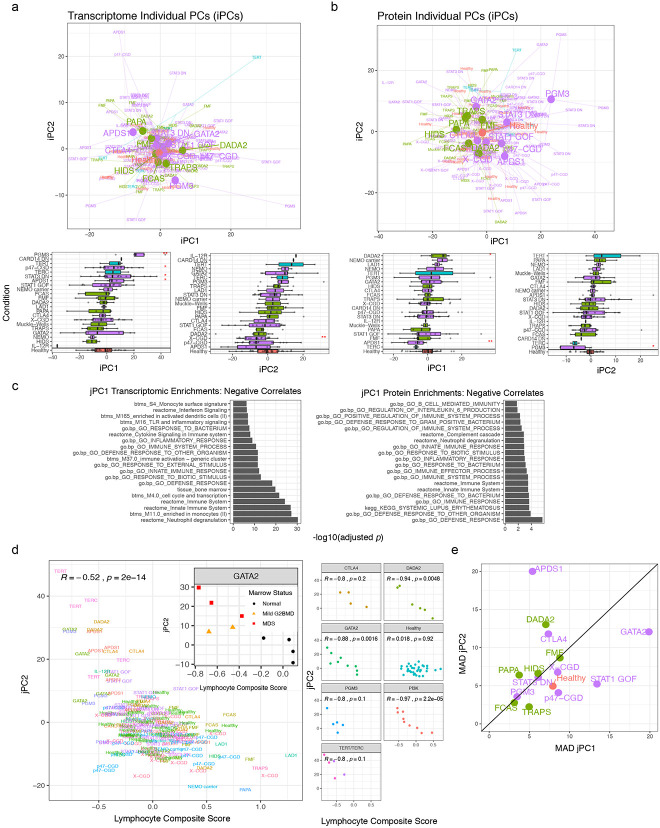
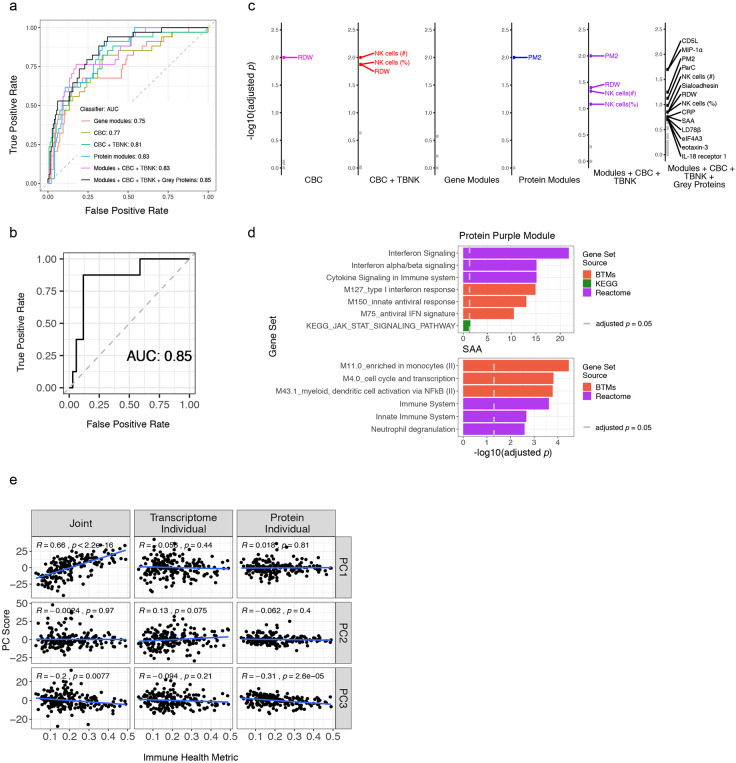
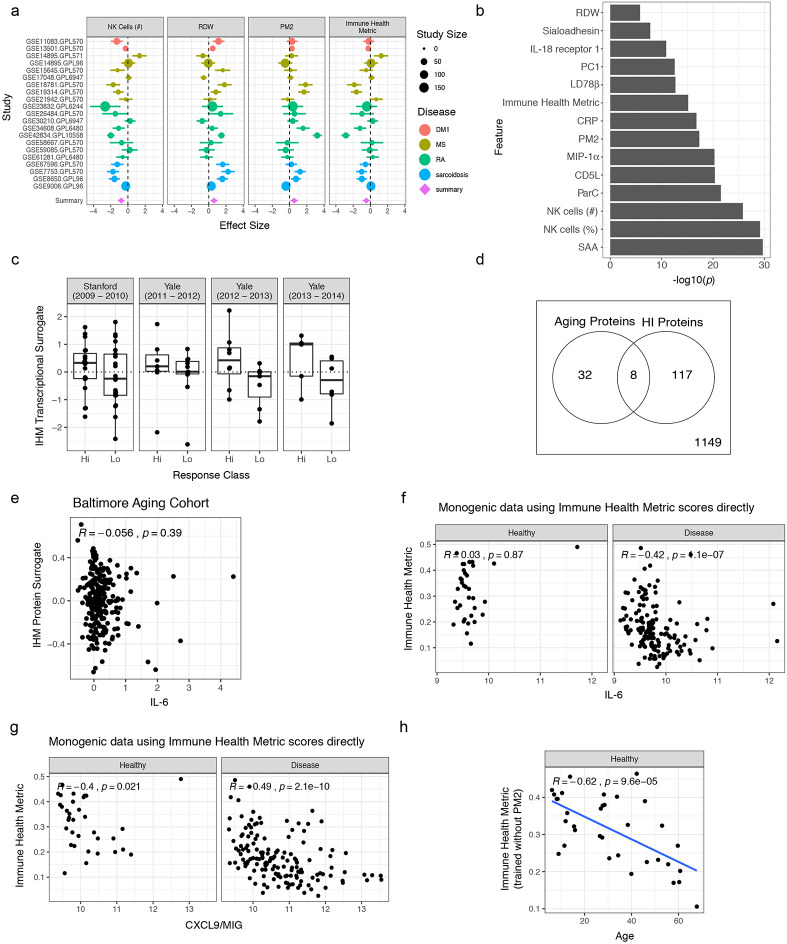
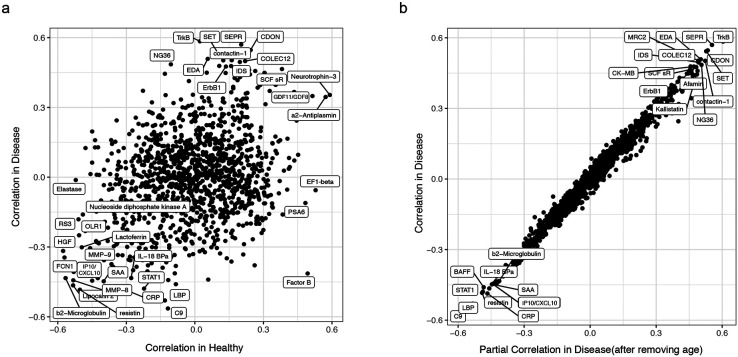
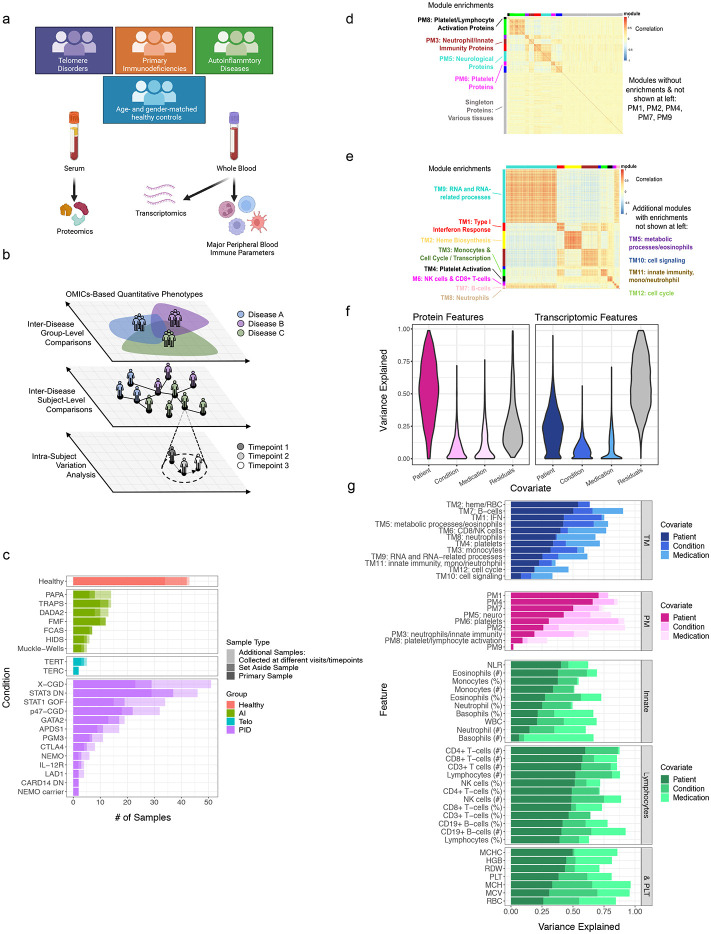
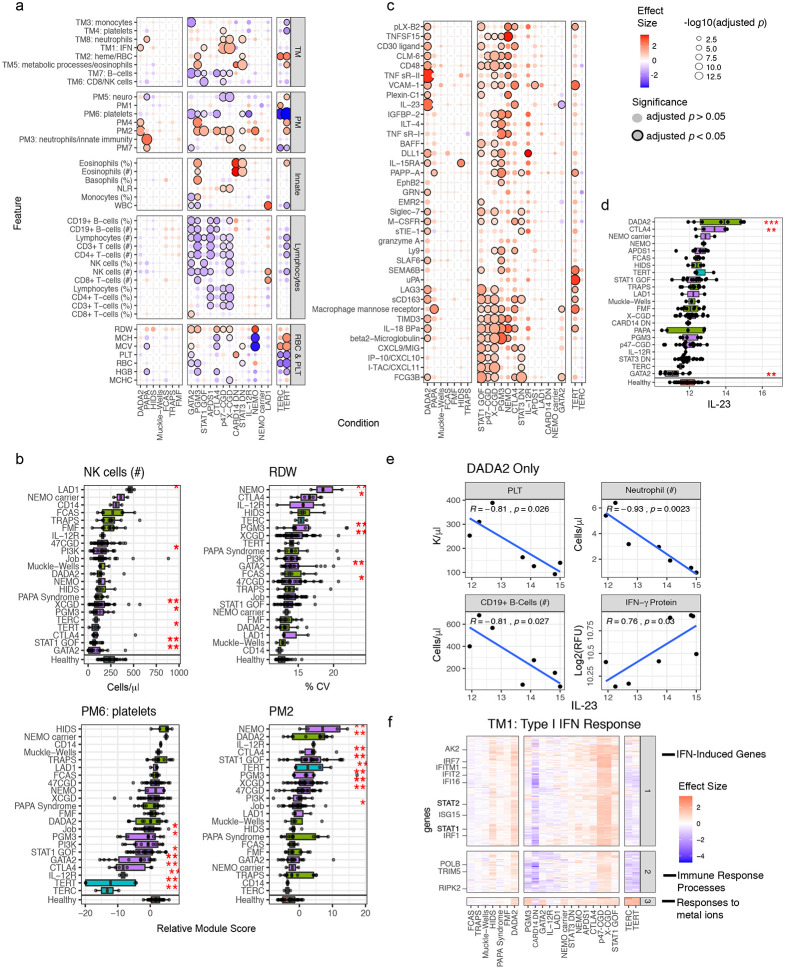
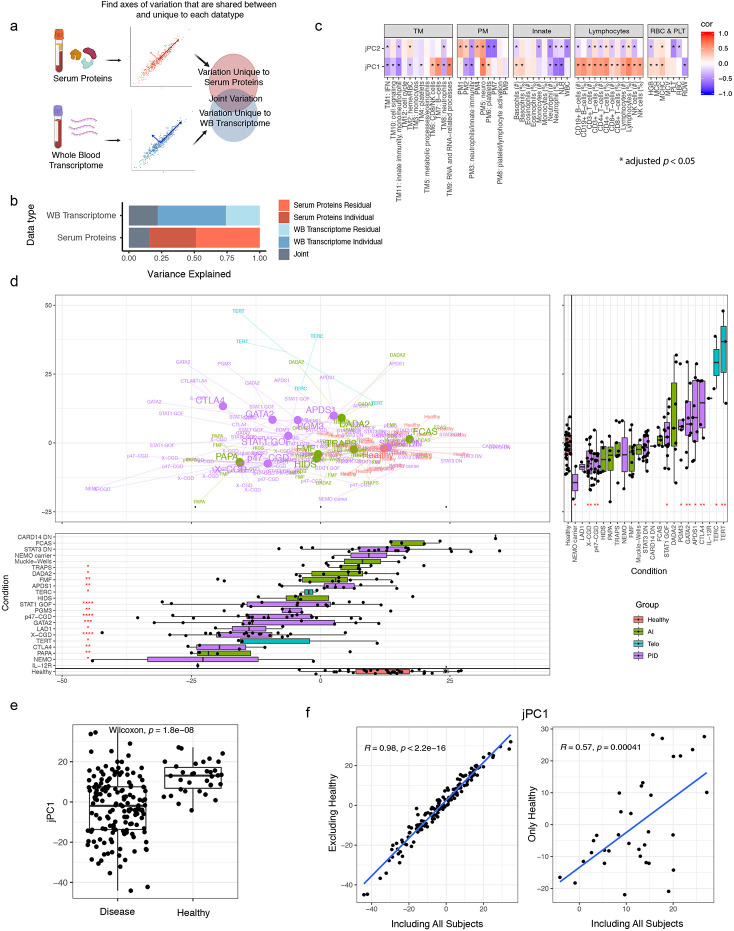
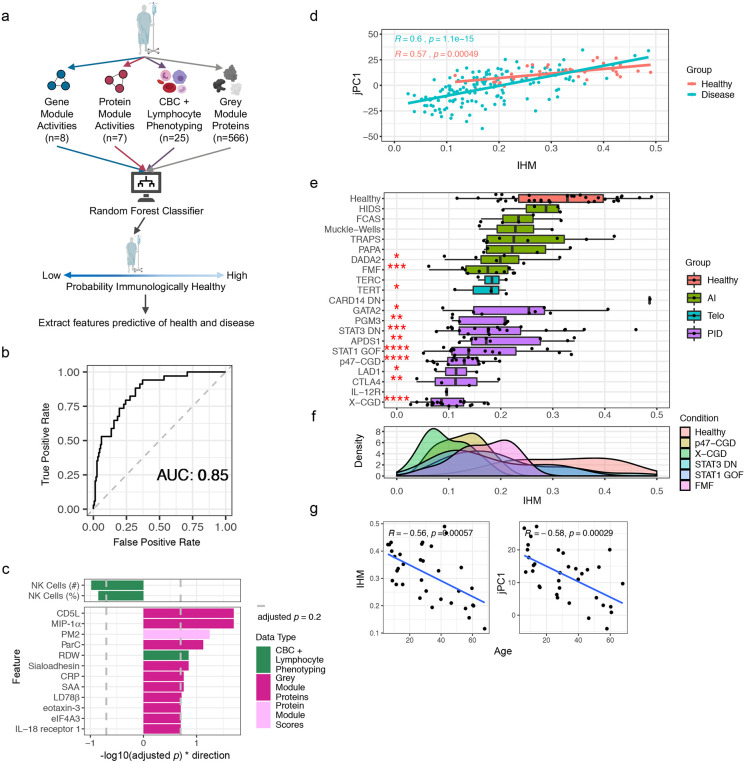
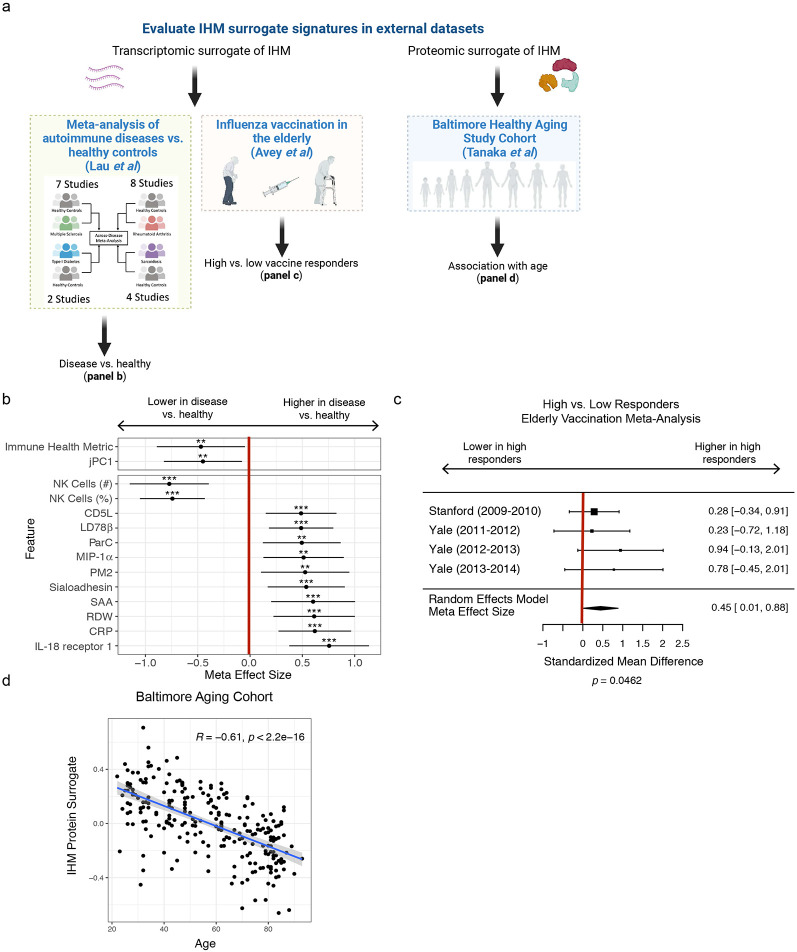
Monogenic diseases are often studied in isolation due to their rarity. Here we utilize multiomics to assess 22 monogenic immune-mediated conditions with age- and sex-matched healthy controls. Despite clearly detectable disease-specific and "pan-disease" signatures, individuals possess stable personal immune states over time. Temporally stable differences among subjects tend to dominate over differences attributable to disease conditions or medication use. Unsupervised principal variation analysis of personal immune states and machine learning classification distinguishing between healthy controls and patients converge to a metric of immune health (IHM). The IHM discriminates healthy from multiple polygenic autoimmune and inflammatory disease states in independent cohorts, marks healthy aging, and is a pre-vaccination predictor of antibody responses to influenza vaccination in the elderly. We identified easy-to-measure circulating protein biomarker surrogates of the IHM that capture immune health variations beyond age. Our work provides a conceptual framework and biomarkers for defining and measuring human immune health.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
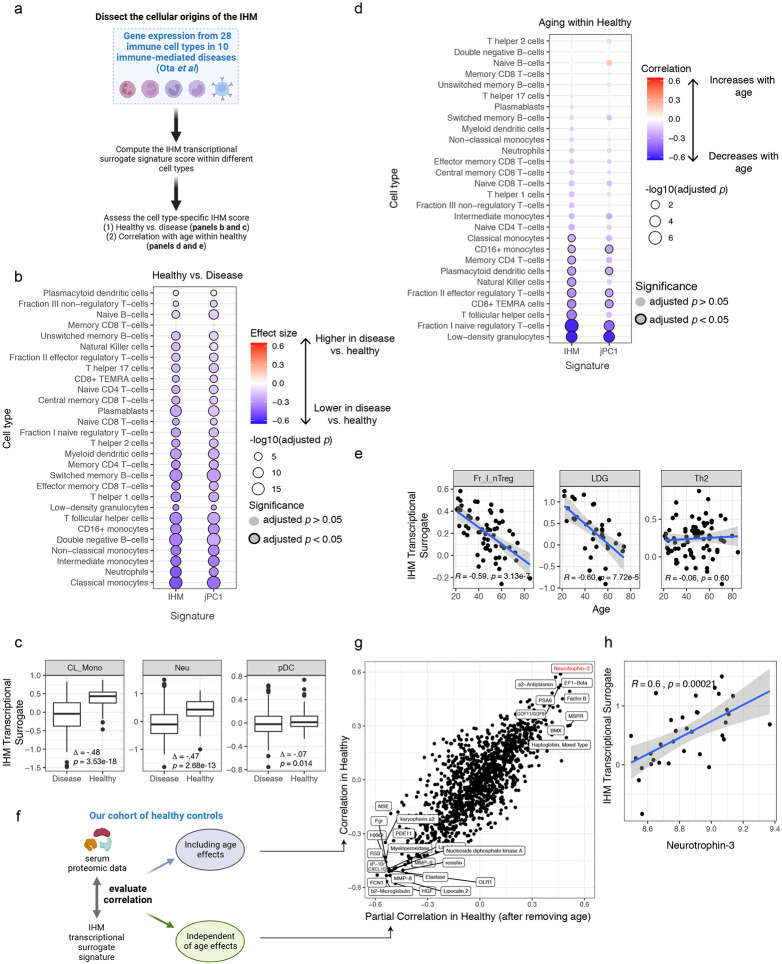
References
-
- Aksentijevich I., and Schnappauf O. (2021). Molecular mechanisms of phenotypic variability in monogenic autoinflammatory diseases. Nat. Rev. Rheumatol. 17, 405–425. - PubMed
-
- Almarza Novoa E., Kasbekar S., Thrasher A.J., Kohn D.B., Sevilla J., Nguyen T., Schwartz J.D., and Bueren J.A. (2018). Leukocyte adhesion deficiency-I: A comprehensive review of all published cases. J. Allergy Clin. Immunol. Pract. 6, 1418–1420.e10. - PubMed
-
- Bergerson J.R.E., and Freeman A.F. (2019). An Update on Syndromes with a Hyper-IgE Phenotype. Immunol. Allergy Clin. North Am. 39, 49–61. - PubMed
Methods References
-
- Templeton A. J. et al. Prognostic Role of Neutrophil-to-Lymphocyte Ratio in Solid Tumors: A Systematic Review and Meta-Analysis. JNCI J. Natl. Cancer Inst. 106, (2014). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
