

This is a preprint.
Inferring compound heterozygosity from large-scale exome sequencing data
- PMID: 36993580
- PMCID: PMC10055215
- DOI: 10.1101/2023.03.19.533370
Inferring compound heterozygosity from large-scale exome sequencing data
Update in
-
Inferring compound heterozygosity from large-scale exome sequencing data.Nat Genet. 2024 Jan;56(1):152-161. doi: 10.1038/s41588-023-01608-3. Epub 2023 Dec 6. Nat Genet. 2024. PMID: 38057443 Free PMC article.
Abstract
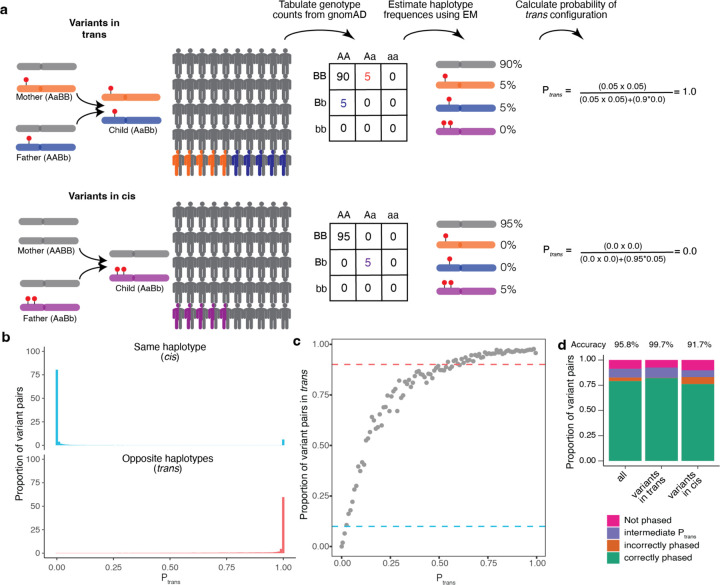
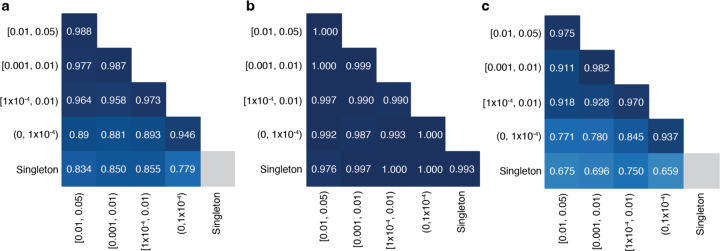
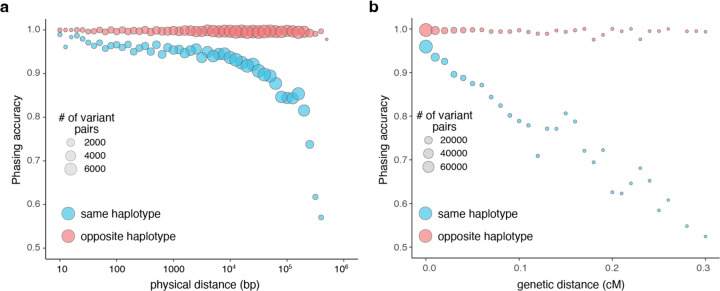
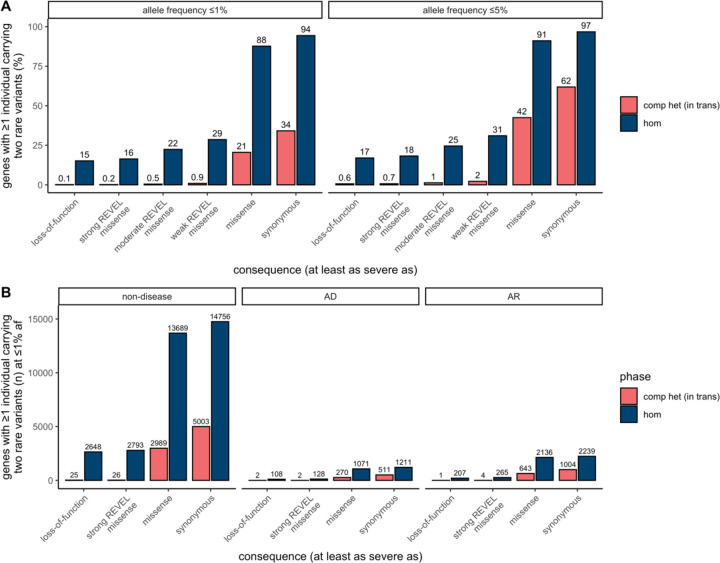
Recessive diseases arise when both the maternal and the paternal copies of a gene are impacted by a damaging genetic variant in the affected individual. When a patient carries two different potentially causal variants in a gene for a given disorder, accurate diagnosis requires determining that these two variants occur on different copies of the chromosome (i.e., are in trans) rather than on the same copy (i.e. in cis). However, current approaches for determining phase, beyond parental testing, are limited in clinical settings. We developed a strategy for inferring phase for rare variant pairs within genes, leveraging genotypes observed in exome sequencing data from the Genome Aggregation Database (gnomAD v2, n=125,748). When applied to trio data where phase can be determined by transmission, our approach estimates phase with 95.7% accuracy and remains accurate even for very rare variants (allele frequency < 1×10-4). We also correctly phase 95.9% of variant pairs in a set of 293 patients with Mendelian conditions carrying presumed causal compound heterozygous variants. We provide a public resource of phasing estimates from gnomAD, including phasing estimates for coding variants across the genome and counts per gene of rare variants in trans, that can aid interpretation of rare co-occurring variants in the context of recessive disease.
Conflict of interest statement
Competing interests B.M.N. is a member of the scientific advisory board at Deep Genomics and Neumora, Inc. (f/k/a RBNC Therapeutics). H.L.R. has received support from Illumina and Microsoft to support rare disease gene discovery and diagnosis. M.J.D. is a founder of Maze Therapeutics and Neumora Therapeutics, Inc. (f/k/a RBNC Therapeutics). A.O.D.L. has consulted for Tome Biosciences and Ono Pharma USA Inc, and is member of the scientific advisory board for Congenica Inc and the Simons Foundation SPARK for Autism study. K.J.K. is a consultant for Tome Biosciences and Vor Biosciences, and a member of the Scientific Advisory Board of Nurture Genomics. D.G.M. is a paid advisor to GlaxoSmithKline, Insitro, Variant Bio and Overtone Therapeutics, and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Google, Merck, Microsoft, Pfizer, and Sanofi-Genzyme. K.E.S. has received support from Microsoft for work related to rare disease diagnostics. The remaining authors declare no competing interests.
Figures


References
-
- McVean G. A. T. et al. The fine-scale structure of recombination rate variation in the human genome. Science 304, 581–584 (2004). - PubMed
-
- Ségurel L., Wyman M. J. & Przeworski M. Determinants of mutation rate variation in the human germline. Annu. Rev. Genomics Hum. Genet. 15, 47–70 (2014). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources