

This is a preprint.
Benchmarking of deep neural networks for predicting personal gene expression from DNA sequence highlights shortcomings
- PMID: 36993652
- PMCID: PMC10055057
- DOI: 10.1101/2023.03.16.532969
Benchmarking of deep neural networks for predicting personal gene expression from DNA sequence highlights shortcomings
Update in
-
Benchmarking of deep neural networks for predicting personal gene expression from DNA sequence highlights shortcomings.Nat Genet. 2023 Dec;55(12):2060-2064. doi: 10.1038/s41588-023-01524-6. Epub 2023 Nov 30. Nat Genet. 2023. PMID: 38036778
Abstract
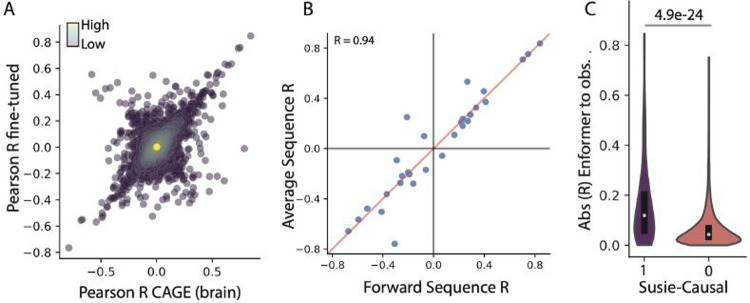
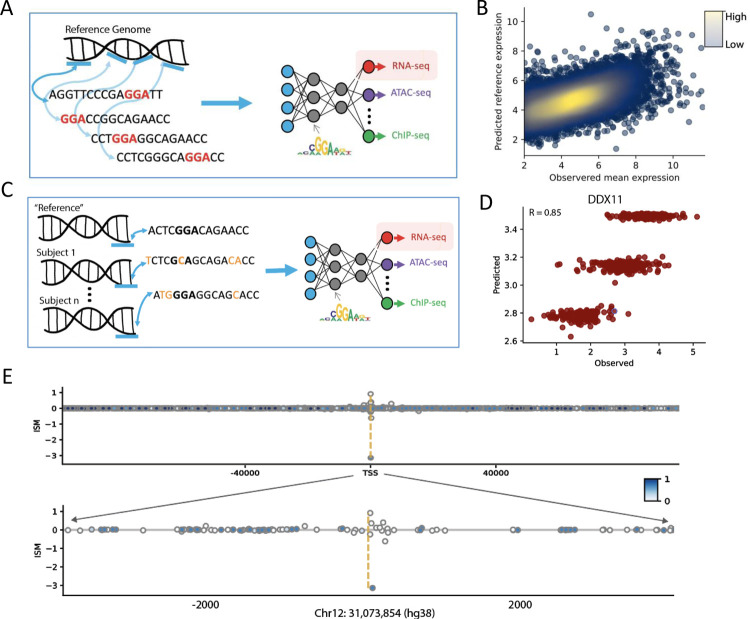
Deep learning methods have recently become the state-of-the-art in a variety of regulatory genomic tasks1-6 including the prediction of gene expression from genomic DNA. As such, these methods promise to serve as important tools in interpreting the full spectrum of genetic variation observed in personal genomes. Previous evaluation strategies have assessed their predictions of gene expression across genomic regions, however, systematic benchmarking is lacking to assess their predictions across individuals, which would directly evaluates their utility as personal DNA interpreters. We used paired Whole Genome Sequencing and gene expression from 839 individuals in the ROSMAP study7 to evaluate the ability of current methods to predict gene expression variation across individuals at varied loci. Our approach identifies a limitation of current methods to correctly predict the direction of variant effects. We show that this limitation stems from insufficiently learnt sequence motif grammar, and suggest new model training strategies to improve performance.
Conflict of interest statement
Competing Interests Statement The authors declare no competing interests.
Figures


References
-
- Eraslan G., Avsec Ž., Gagneur J. & Theis F. J. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 20, 389–403 (2019). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials