

Rare Single Nucleotide and Copy Number Variants and the Etiology of Congenital Obstructive Uropathy: Implications for Genetic Diagnosis
- PMID: 36995132
- PMCID: PMC10278788
- DOI: 10.1681/ASN.0000000000000132
Rare Single Nucleotide and Copy Number Variants and the Etiology of Congenital Obstructive Uropathy: Implications for Genetic Diagnosis
Abstract
Significance statement: Congenital obstructive uropathy (COU) is a prevalent human developmental defect with highly heterogeneous clinical presentations and outcomes. Genetics may refine diagnosis, prognosis, and treatment, but the genomic architecture of COU is largely unknown. Comprehensive genomic screening study of 733 cases with three distinct COU subphenotypes revealed disease etiology in 10.0% of them. We detected no significant differences in the overall diagnostic yield among COU subphenotypes, with characteristic variable expressivity of several mutant genes. Our findings therefore may legitimize a genetic first diagnostic approach for COU, especially when burdening clinical and imaging characterization is not complete or available.
Background: Congenital obstructive uropathy (COU) is a common cause of developmental defects of the urinary tract, with heterogeneous clinical presentation and outcome. Genetic analysis has the potential to elucidate the underlying diagnosis and help risk stratification.
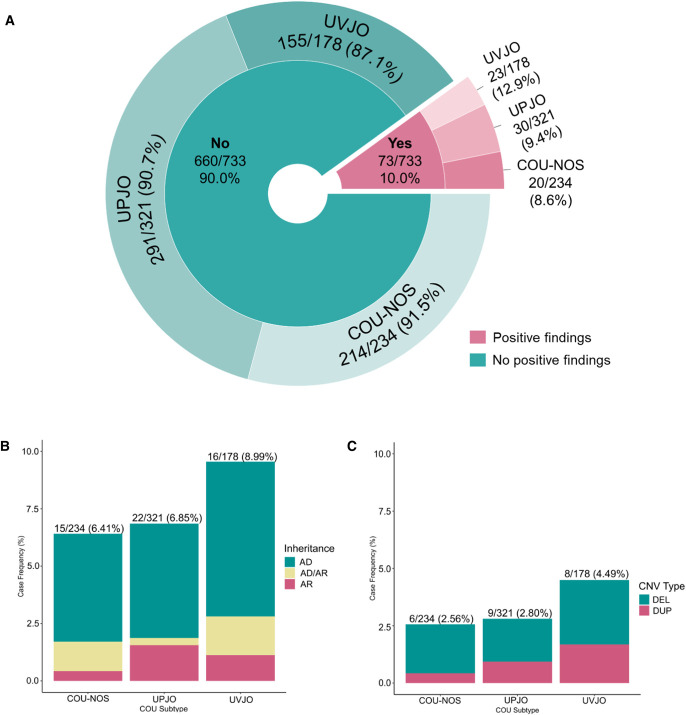
Methods: We performed a comprehensive genomic screen of 733 independent COU cases, which consisted of individuals with ureteropelvic junction obstruction ( n =321), ureterovesical junction obstruction/congenital megaureter ( n =178), and COU not otherwise specified (COU-NOS; n =234).
Results: We identified pathogenic single nucleotide variants (SNVs) in 53 (7.2%) cases and genomic disorders (GDs) in 23 (3.1%) cases. We detected no significant differences in the overall diagnostic yield between COU sub-phenotypes, and pathogenic SNVs in several genes were associated to any of the three categories. Hence, although COU may appear phenotypically heterogeneous, COU phenotypes are likely to share common molecular bases. On the other hand, mutations in TNXB were more often identified in COU-NOS cases, demonstrating the diagnostic challenge in discriminating COU from hydronephrosis secondary to vesicoureteral reflux, particularly when diagnostic imaging is incomplete. Pathogenic SNVs in only six genes were found in more than one individual, supporting high genetic heterogeneity. Finally, convergence between data on SNVs and GDs suggest MYH11 as a dosage-sensitive gene possibly correlating with severity of COU.
Conclusions: We established a genomic diagnosis in 10.0% of COU individuals. The findings underscore the urgent need to identify novel genetic susceptibility factors to COU to better define the natural history of the remaining 90% of cases without a molecular diagnosis.
Copyright © 2023 by the American Society of Nephrology.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
