

Adeno-associated virus type 2 in US children with acute severe hepatitis
- PMID: 36996871
- PMCID: PMC10170441
- DOI: 10.1038/s41586-023-05949-1
Adeno-associated virus type 2 in US children with acute severe hepatitis
Abstract
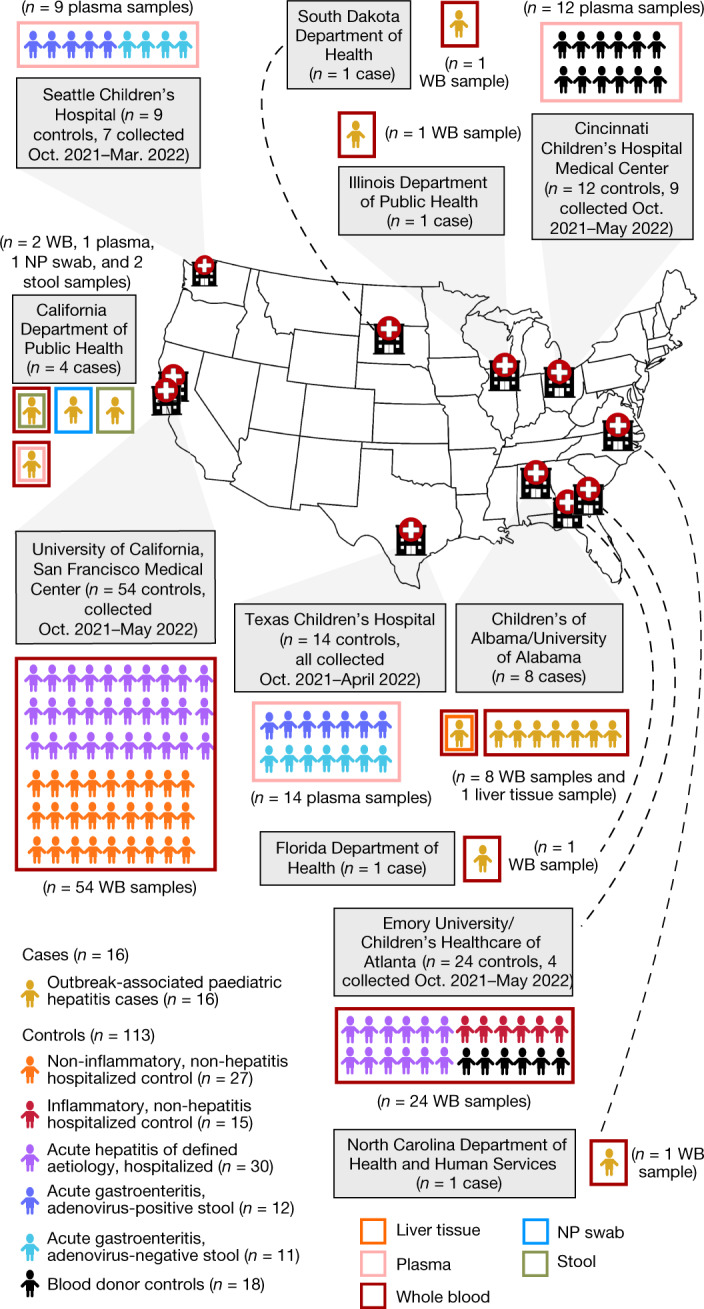
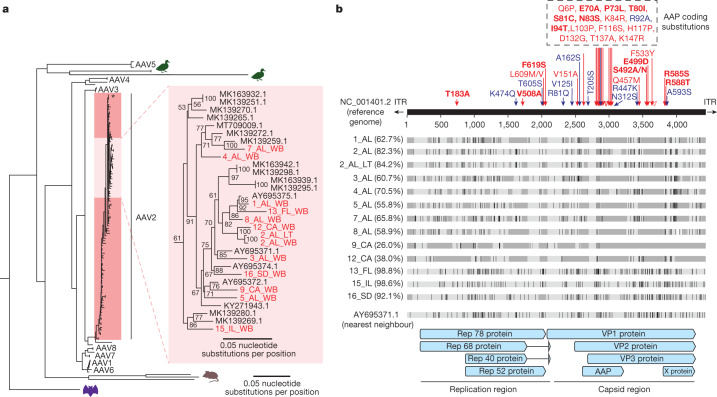
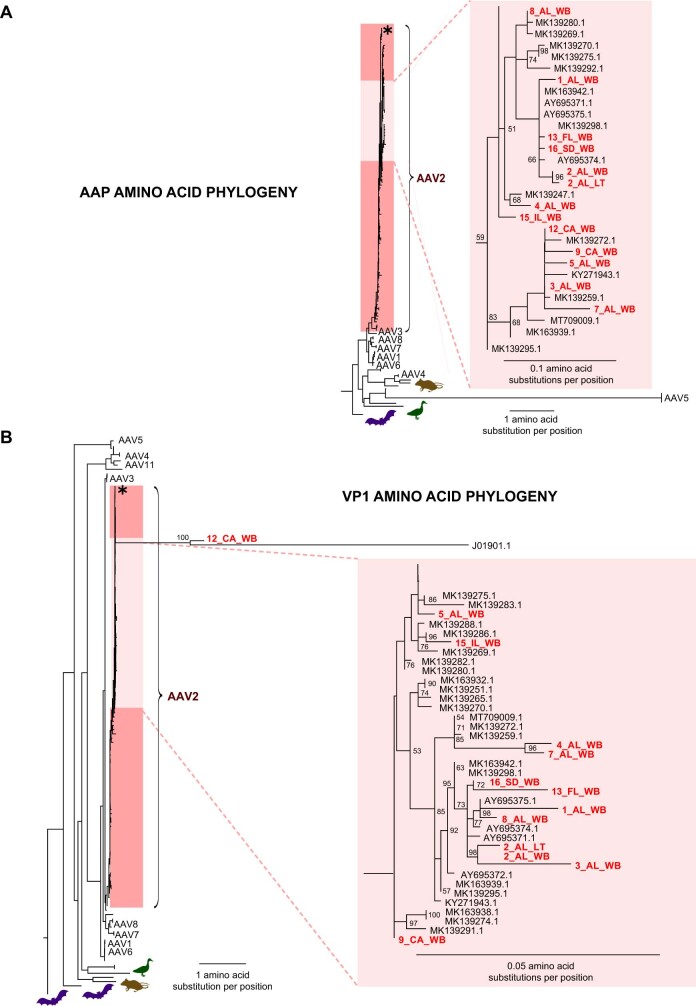
As of August 2022, clusters of acute severe hepatitis of unknown aetiology in children have been reported from 35 countries, including the USA1,2. Previous studies have found human adenoviruses (HAdVs) in the blood from patients in Europe and the USA3-7, although it is unclear whether this virus is causative. Here we used PCR testing, viral enrichment-based sequencing and agnostic metagenomic sequencing to analyse samples from 16 HAdV-positive cases from 1 October 2021 to 22 May 2022, in parallel with 113 controls. In blood from 14 cases, adeno-associated virus type 2 (AAV2) sequences were detected in 93% (13 of 14), compared to 4 (3.5%) of 113 controls (P < 0.001) and to 0 of 30 patients with hepatitis of defined aetiology (P < 0.001). In controls, HAdV type 41 was detected in blood from 9 (39.1%) of the 23 patients with acute gastroenteritis (without hepatitis), including 8 of 9 patients with positive stool HAdV testing, but co-infection with AAV2 was observed in only 3 (13.0%) of these 23 patients versus 93% of cases (P < 0.001). Co-infections by Epstein-Barr virus, human herpesvirus 6 and/or enterovirus A71 were also detected in 12 (85.7%) of 14 cases, with higher herpesvirus detection in cases versus controls (P < 0.001). Our findings suggest that the severity of the disease is related to co-infections involving AAV2 and one or more helper viruses.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
C.Y.C. is a founder of Delve Bio and on the scientific advisory board for Delve Bio, Mammoth Biosciences, BiomeSense and Poppy Health. He is also a co-inventor on US patent 11380421, “Pathogen detection using next generation sequencing”, under which algorithms for taxonomic classification, filtering and pathogen detection are used by SURPI+ software. C.Y.C. receives research support unrelated to this manuscript from Abbott Laboratories, Inc. The institution of C.A.R. has received financial support to conduct clinical research unrelated to this manuscript from BioFire Inc., GSK, Janssen, MedImmune, Merck, Moderna, Novavax, PaxVax, Pfizer, Regeneron and Sanofi-Pasteur. She is co-inventor of patented respiratory syncytial virus vaccine technology, which has been licensed to Meissa Vaccines, Inc. K.S.G. receives research support unrelated to this manuscript from Thermo-Fisher and has a royalty-generating collaborative agreement with Zeptometrix. The other authors declare no competing interests.
Figures

Comment in
-
Severe hepatitis outbreak in children linked to AAV2 virus.Nature. 2023 May;617(7961):471-472. doi: 10.1038/d41586-023-00570-8. Nature. 2023. PMID: 36997704 No abstract available.
References
-
- Severe acute hepatitis of unknown aetiology in children - multi-country. WHO Disease Outbreak Newshttps://www.who.int/emergencies/disease-outbreak-news/item/2022-DON400 (2022).
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
