

Functional characterization of human genomic variation linked to polygenic diseases
- PMID: 36997428
- PMCID: PMC11025698
- DOI: 10.1016/j.tig.2023.02.014
Functional characterization of human genomic variation linked to polygenic diseases
Abstract
The burden of human disease lies predominantly in polygenic diseases. Since the early 2000s, genome-wide association studies (GWAS) have identified genetic variants and loci associated with complex traits. These have ranged from variants in coding sequences to mutations in regulatory regions, such as promoters and enhancers, as well as mutations affecting mediators of mRNA stability and other downstream regulators, such as 5' and 3'-untranslated regions (UTRs), long noncoding RNA (lncRNA), and miRNA. Recent research advances in genetics have utilized a combination of computational techniques, high-throughput in vitro and in vivo screening modalities, and precise genome editing to impute the function of diverse classes of genetic variants identified through GWAS. In this review, we highlight the vastness of genomic variants associated with polygenic disease risk and address recent advances in how genetic tools can be used to functionally characterize them.
Keywords: CRISPR; GWAS variants; MPRA; chromatin capture; colocalization.
Copyright © 2023 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests No interests are declared.
Figures
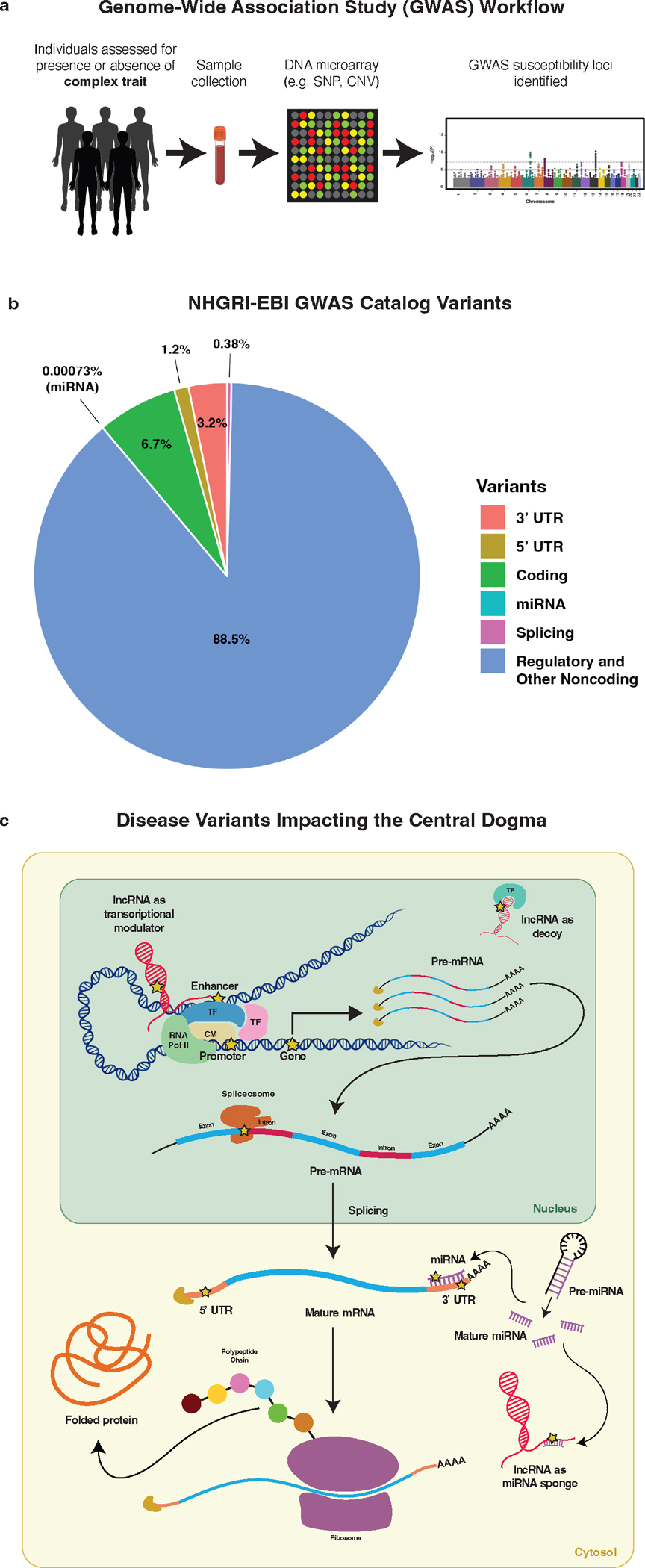
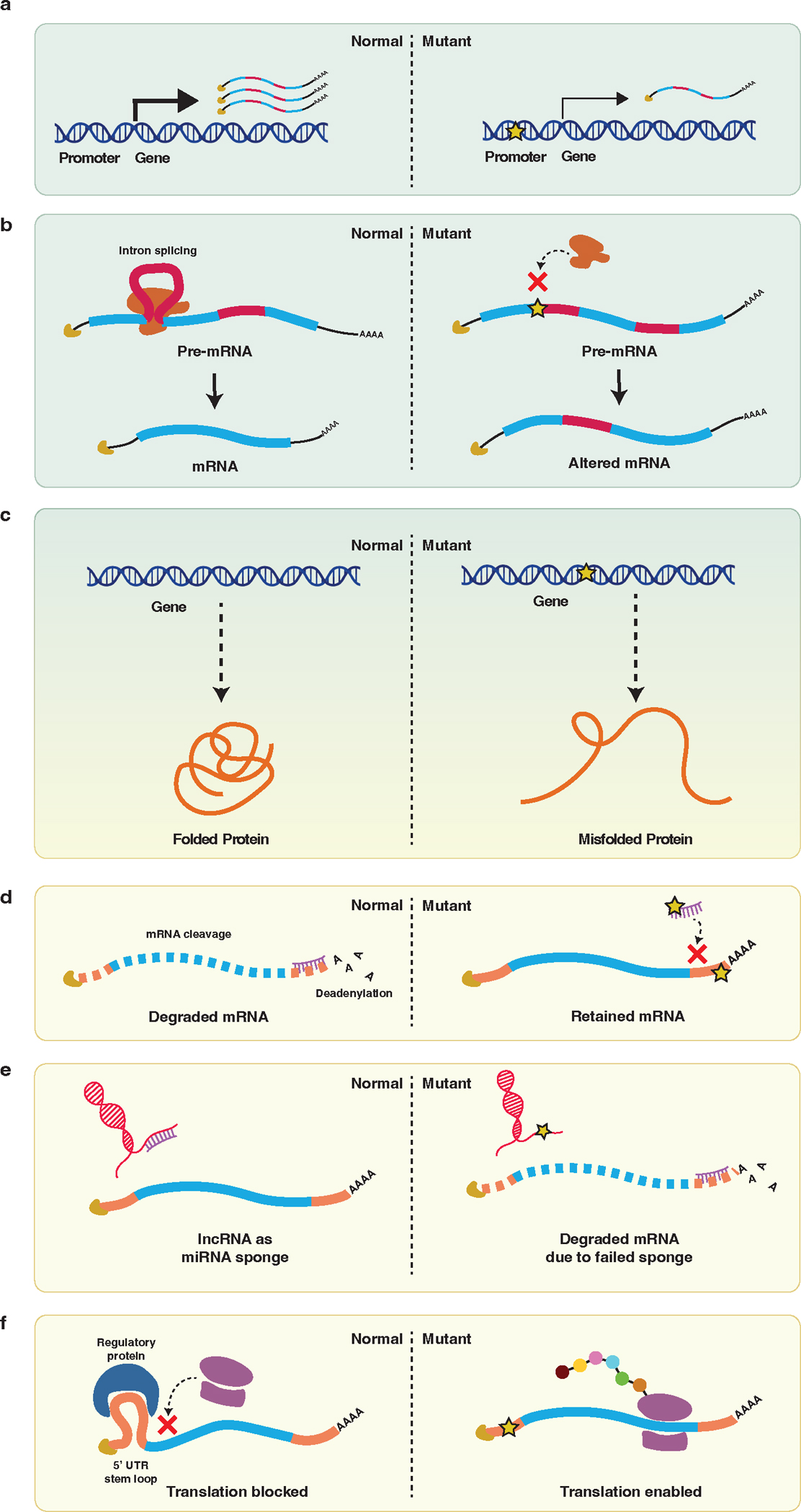
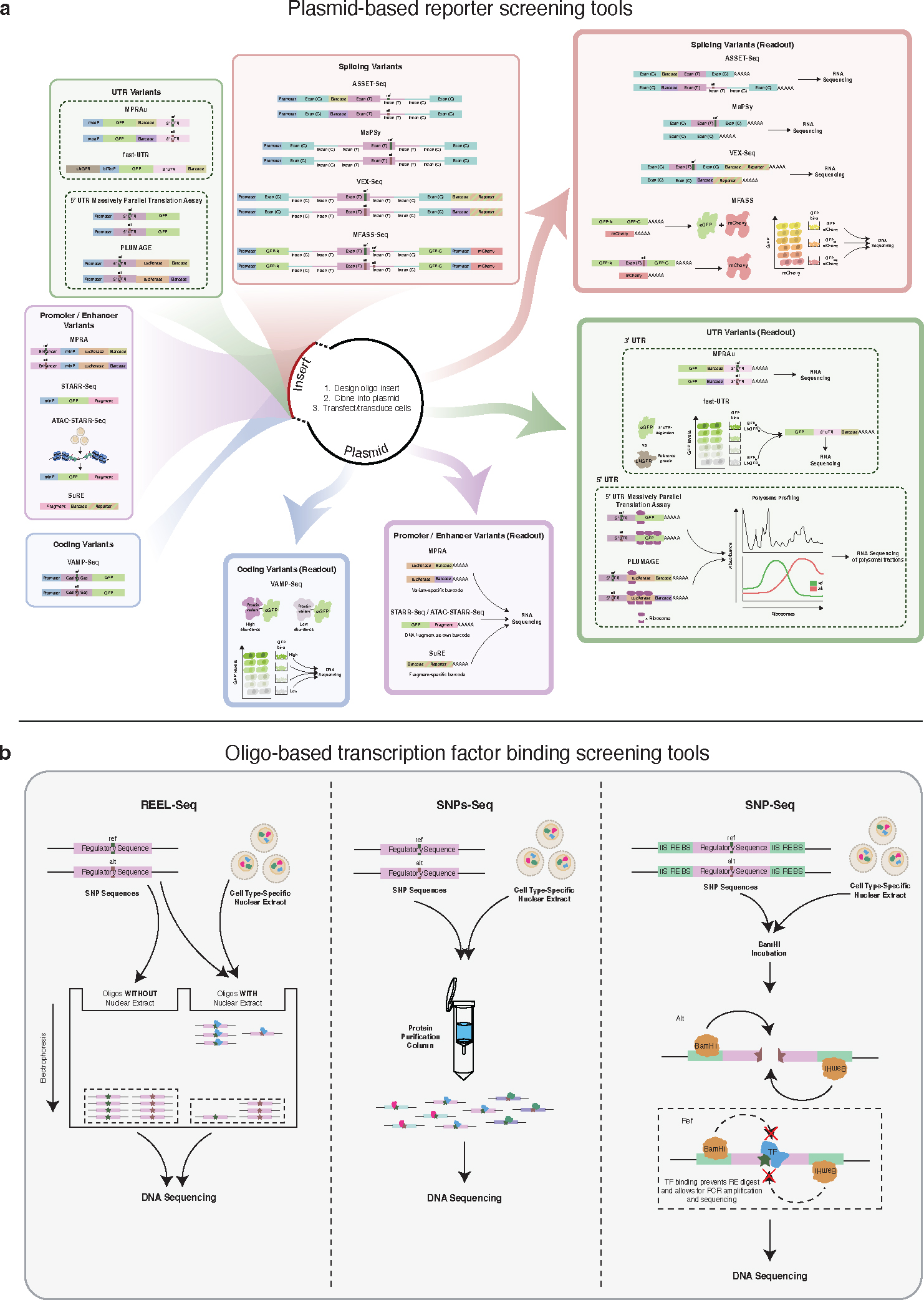
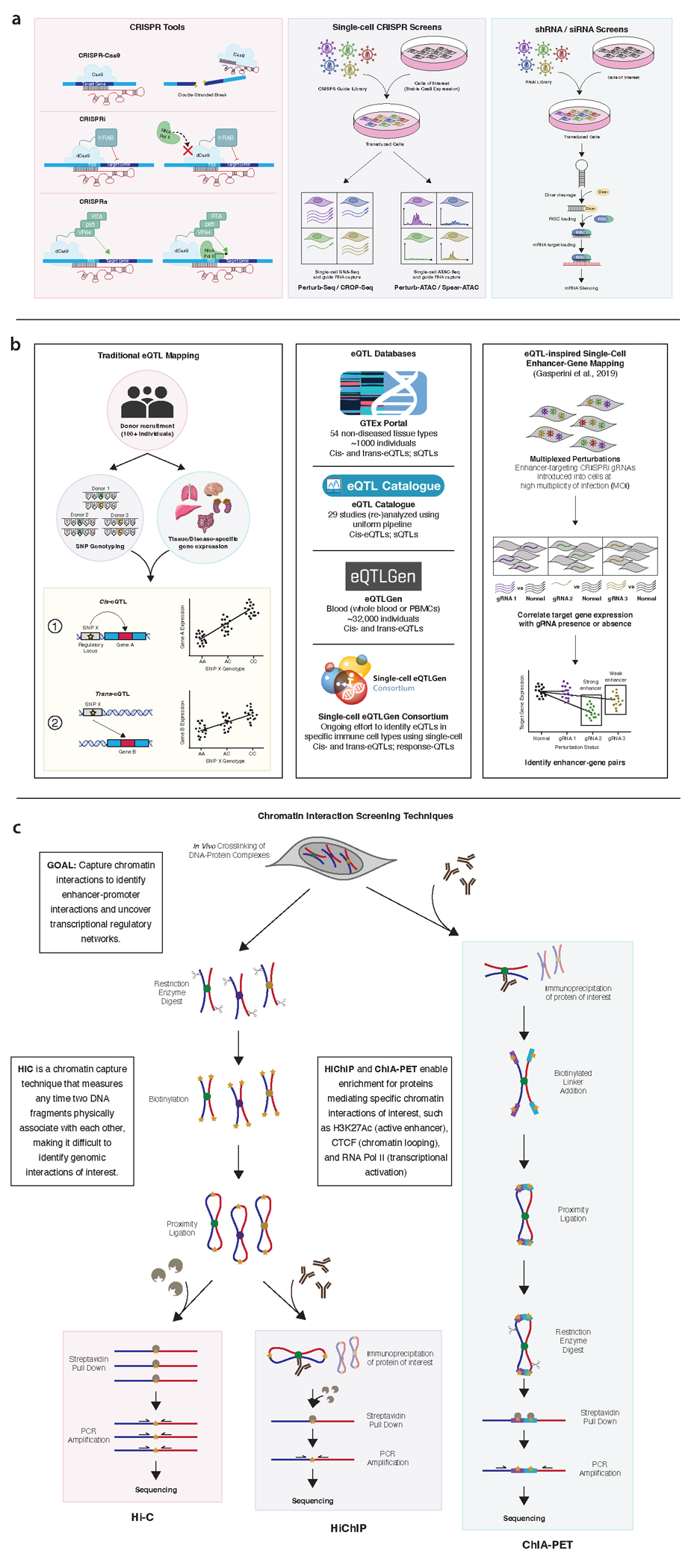
References
-
- Glazier AM et al. (2002) Finding Genes That Underlie Complex Traits. Science 298, 2345–2349 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
