

Precision-activated T-cell engagers targeting HER2 or EGFR and CD3 mitigate on-target, off-tumor toxicity for immunotherapy in solid tumors
- PMID: 36997747
- PMCID: PMC10132983
- DOI: 10.1038/s43018-023-00536-9
Precision-activated T-cell engagers targeting HER2 or EGFR and CD3 mitigate on-target, off-tumor toxicity for immunotherapy in solid tumors
Abstract
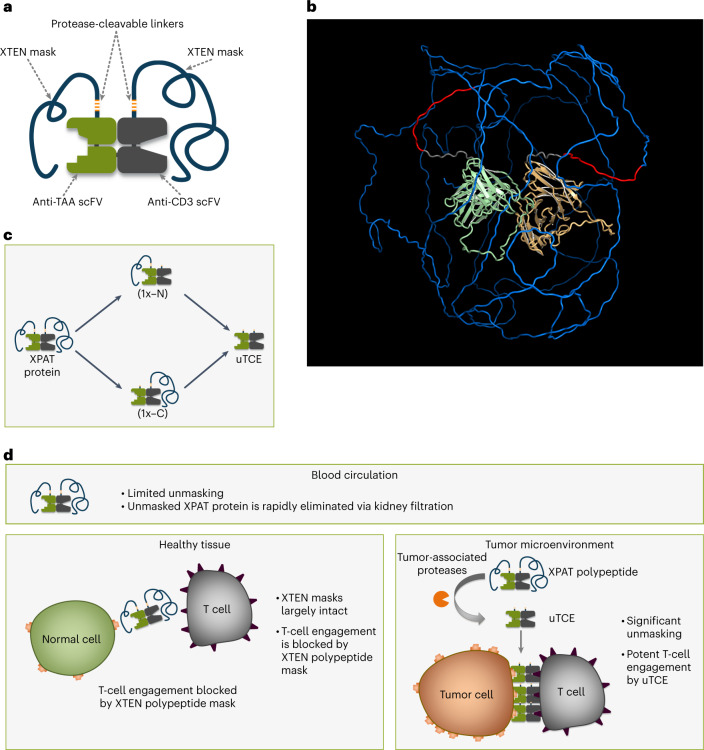
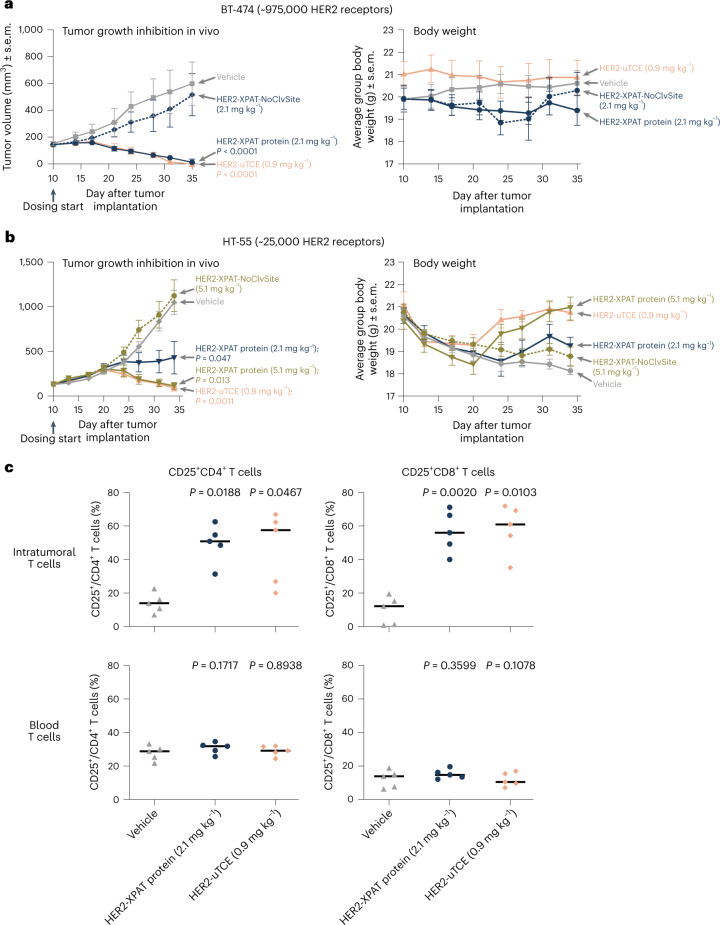
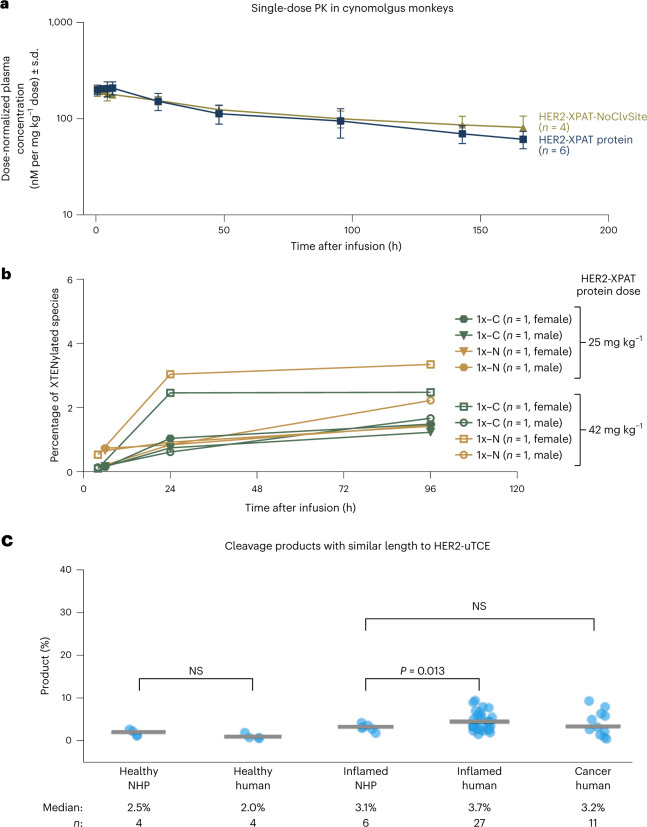
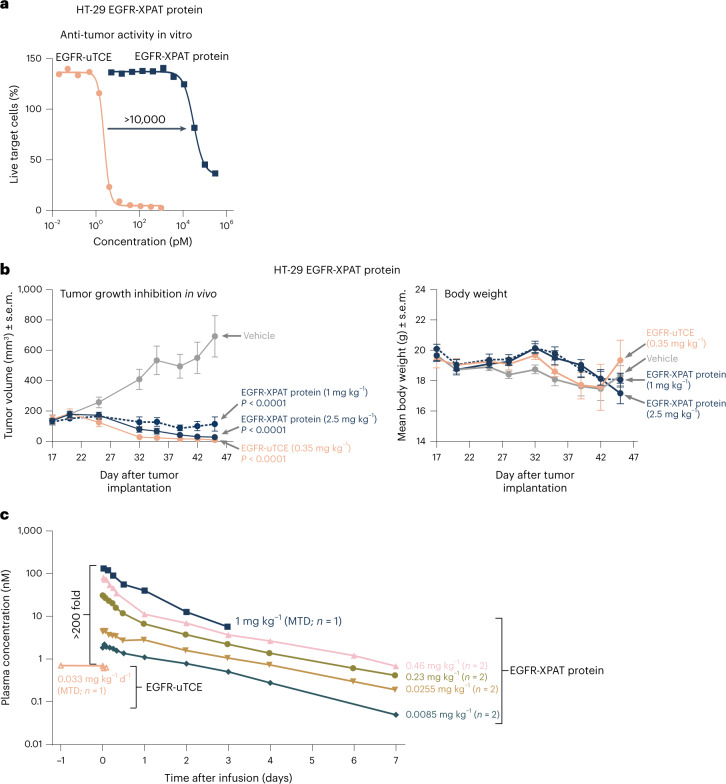
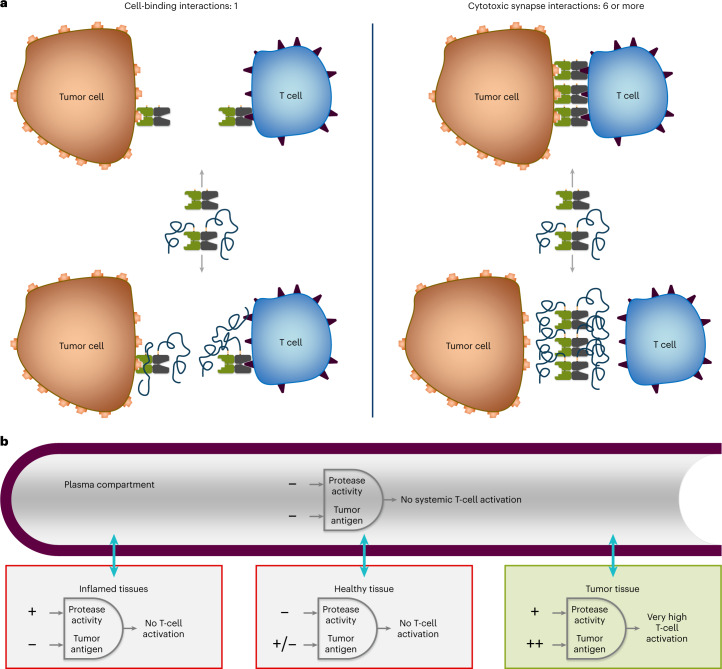
To enhance the therapeutic index of T-cell engagers (TCEs), we engineered masked, precision-activated TCEs (XPAT proteins), targeting a tumor antigen (human epidermal growth factor receptor 2 (HER2) or epidermal growth factor receptor (EGFR)) and CD3. Unstructured XTEN polypeptide masks flank the N and C termini of the TCE and are designed to be released by proteases in the tumor microenvironment. In vitro, unmasked HER2-XPAT (uTCE) demonstrates potent cytotoxicity, with XTEN polypeptide masking providing up to 4-log-fold protection. In vivo, HER2-XPAT protein induces protease-dependent antitumor activity and is proteolytically stable in healthy tissues. In non-human primates, HER2-XPAT protein demonstrates a strong safety margin (>400-fold increase in tolerated maximum concentration versus uTCE). HER2-XPAT protein cleavage is low and similar in plasma samples from healthy and diseased humans and non-human primates, supporting translatability of stability to patients. EGFR-XPAT protein confirmed the utility of XPAT technology for tumor targets more widely expressed in healthy tissues.
© 2023. The Author(s).
Conflict of interest statement
All authors are employees of and own stock in Amunix Pharmaceuticals (a Sanofi company). This study was funded by Amunix Pharmaceuticals.
Figures






Comment in
-
Masking T cell engagers mitigates on-target off-tumor activity.Nat Cancer. 2023 Apr;4(4):439-441. doi: 10.1038/s43018-023-00529-8. Nat Cancer. 2023. PMID: 36997746 No abstract available.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
