

No evidence for a common blood microbiome based on a population study of 9,770 healthy humans
- PMID: 36997797
- PMCID: PMC10159858
- DOI: 10.1038/s41564-023-01350-w
No evidence for a common blood microbiome based on a population study of 9,770 healthy humans
Abstract
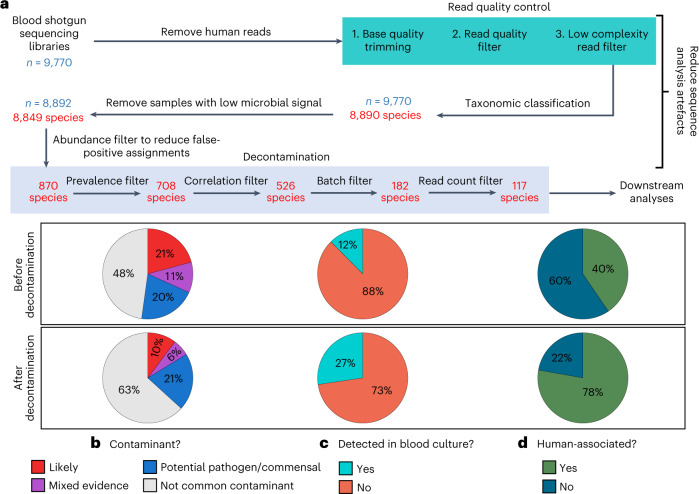
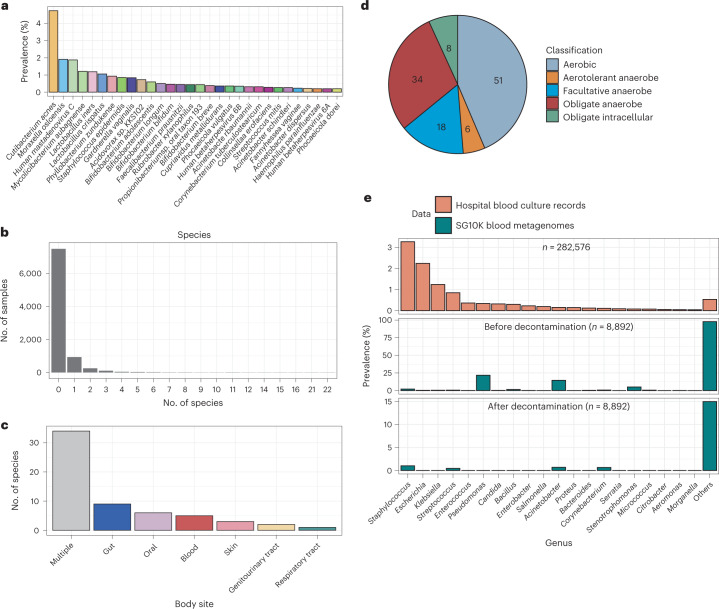
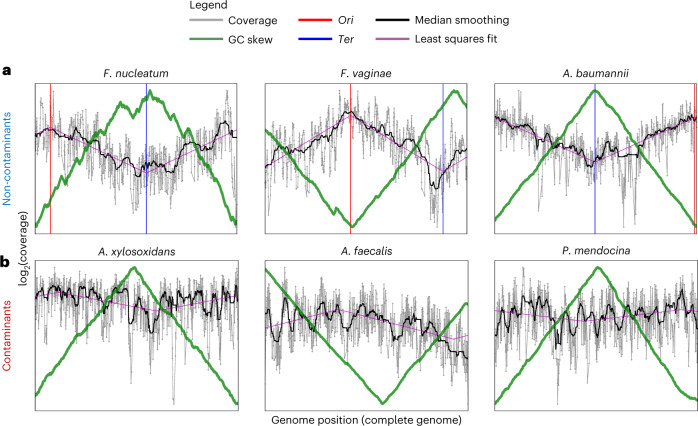
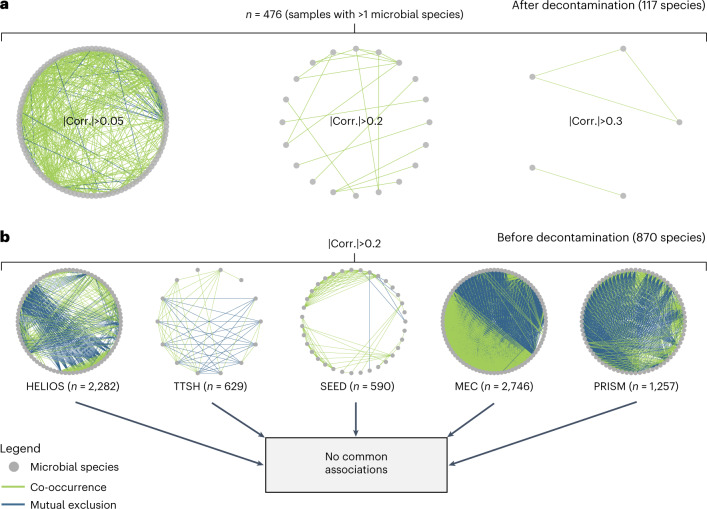
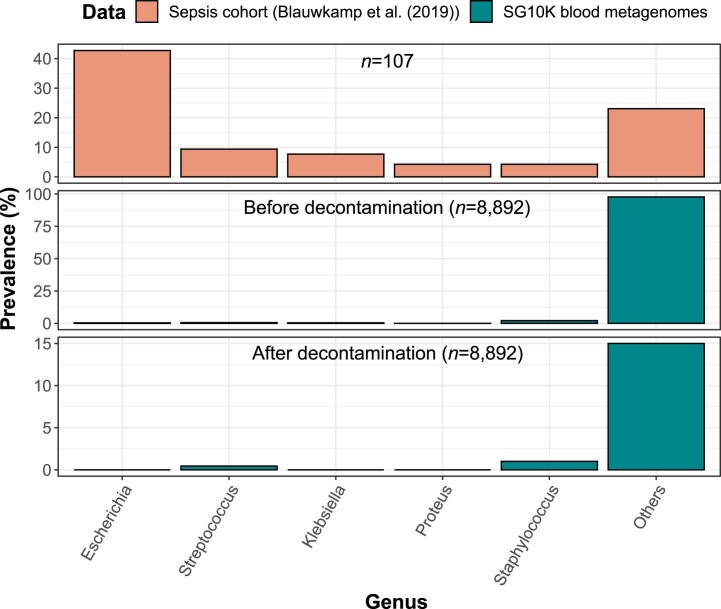
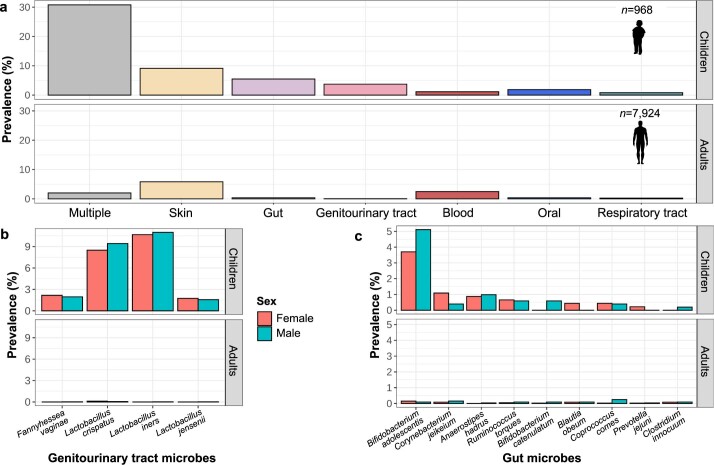
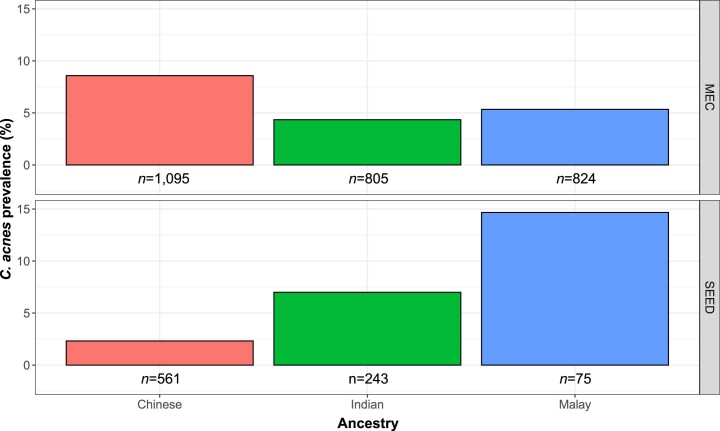
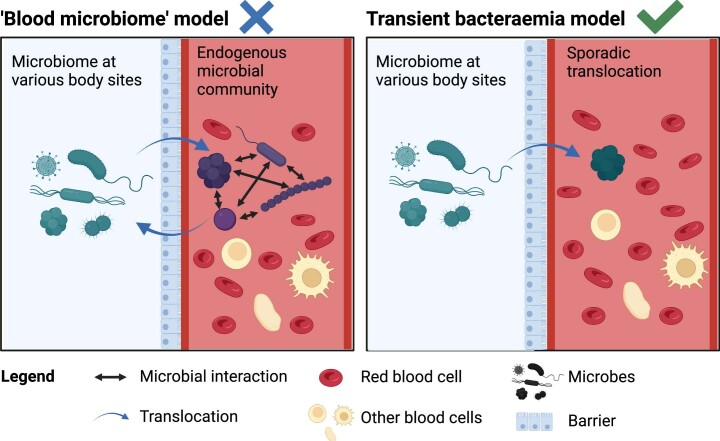
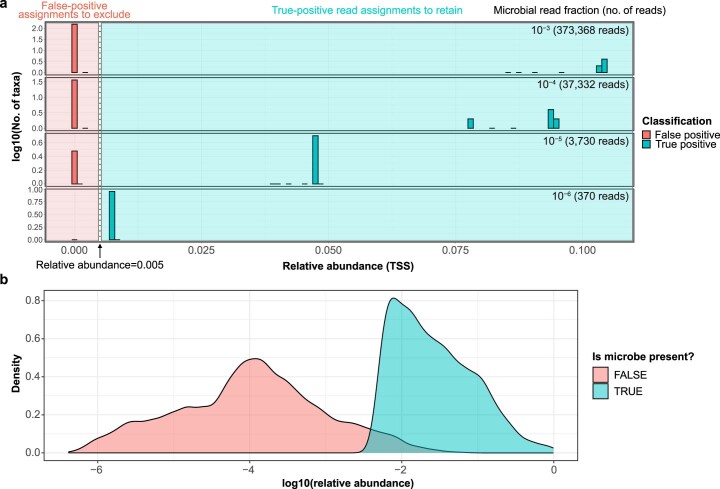
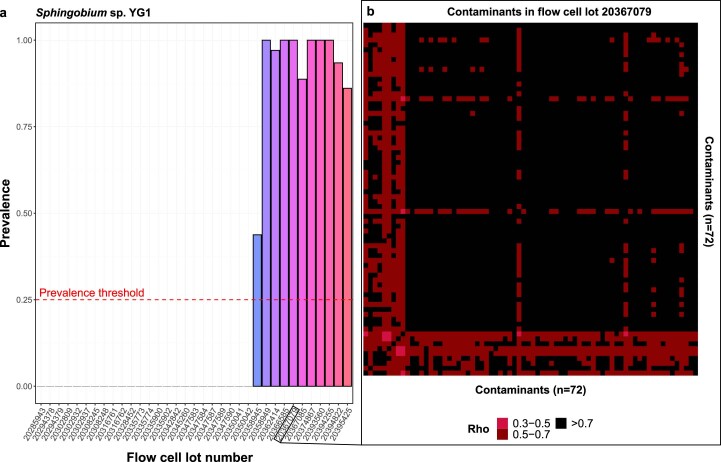
Human blood is conventionally considered sterile but recent studies suggest the presence of a blood microbiome in healthy individuals. Here we characterized the DNA signatures of microbes in the blood of 9,770 healthy individuals using sequencing data from multiple cohorts. After filtering for contaminants, we identified 117 microbial species in blood, some of which had DNA signatures of microbial replication. They were primarily commensals associated with the gut (n = 40), mouth (n = 32) and genitourinary tract (n = 18), and were distinct from pathogens detected in hospital blood cultures. No species were detected in 84% of individuals, while the remainder only had a median of one species. Less than 5% of individuals shared the same species, no co-occurrence patterns between different species were observed and no associations between host phenotypes and microbes were found. Overall, these results do not support the hypothesis of a consistent core microbiome endogenous to human blood. Rather, our findings support the transient and sporadic translocation of commensal microbes from other body sites into the bloodstream.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
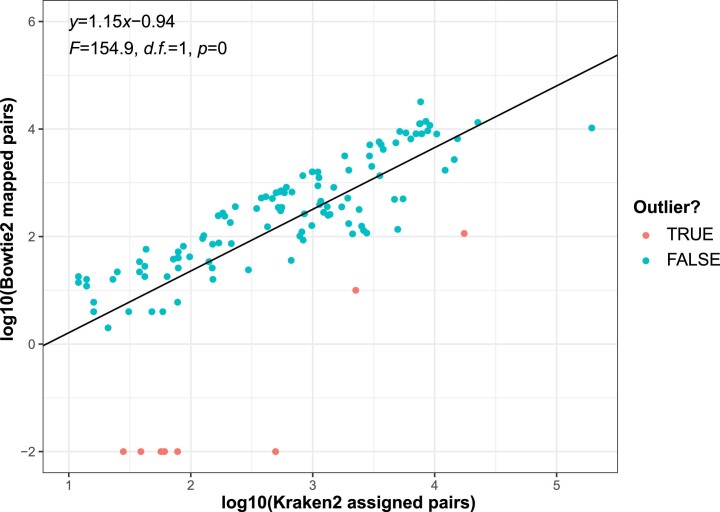
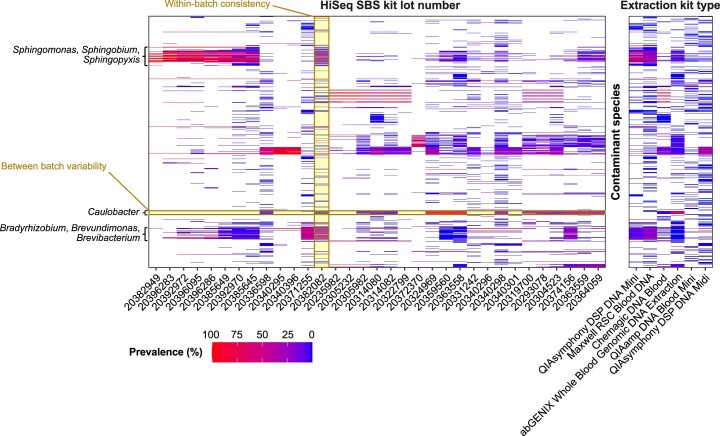
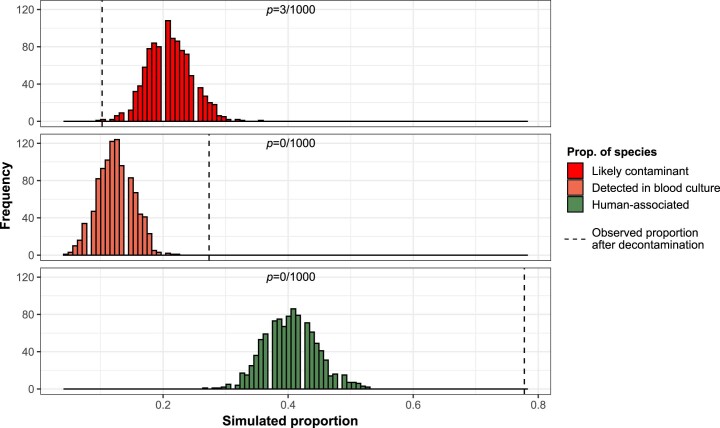
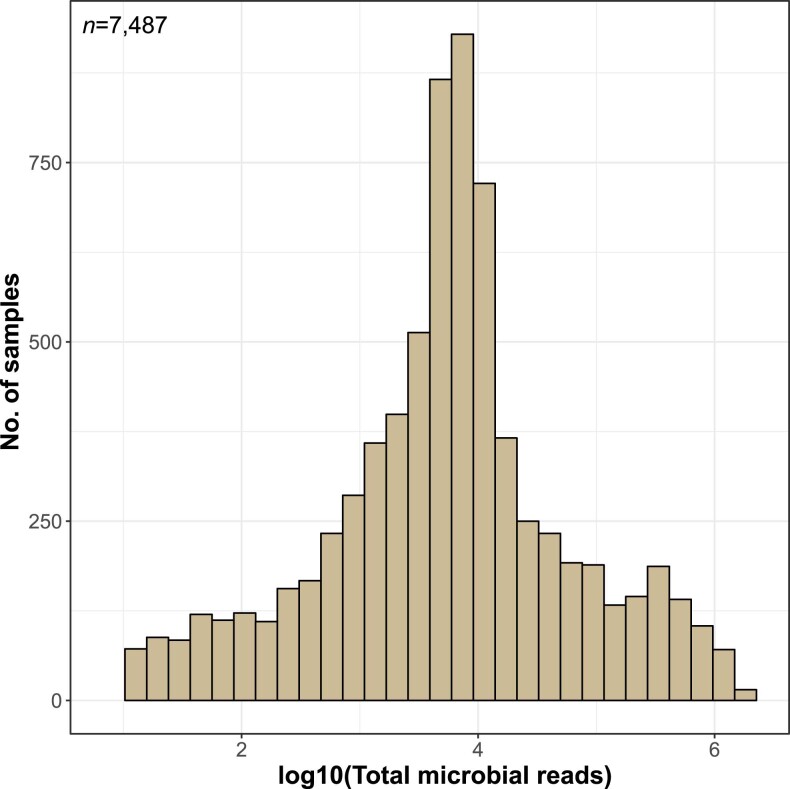
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
