

CDK9 inhibition induces epigenetic reprogramming revealing strategies to circumvent resistance in lymphoma
- PMID: 36998071
- PMCID: PMC10061728
- DOI: 10.1186/s12943-023-01762-6
CDK9 inhibition induces epigenetic reprogramming revealing strategies to circumvent resistance in lymphoma
Abstract
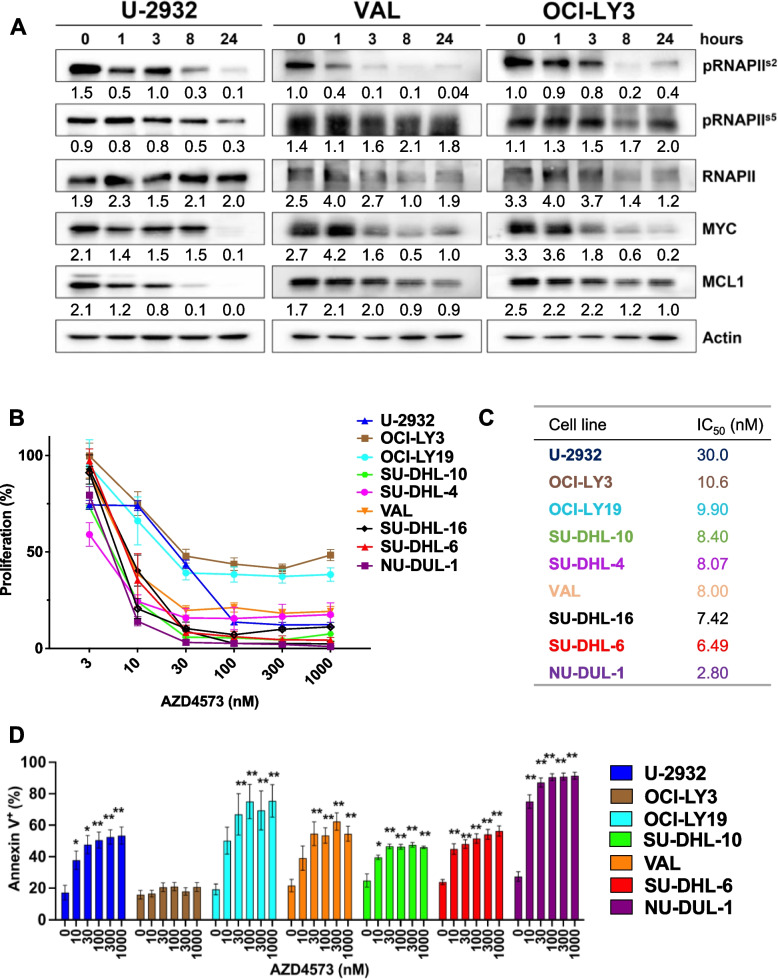
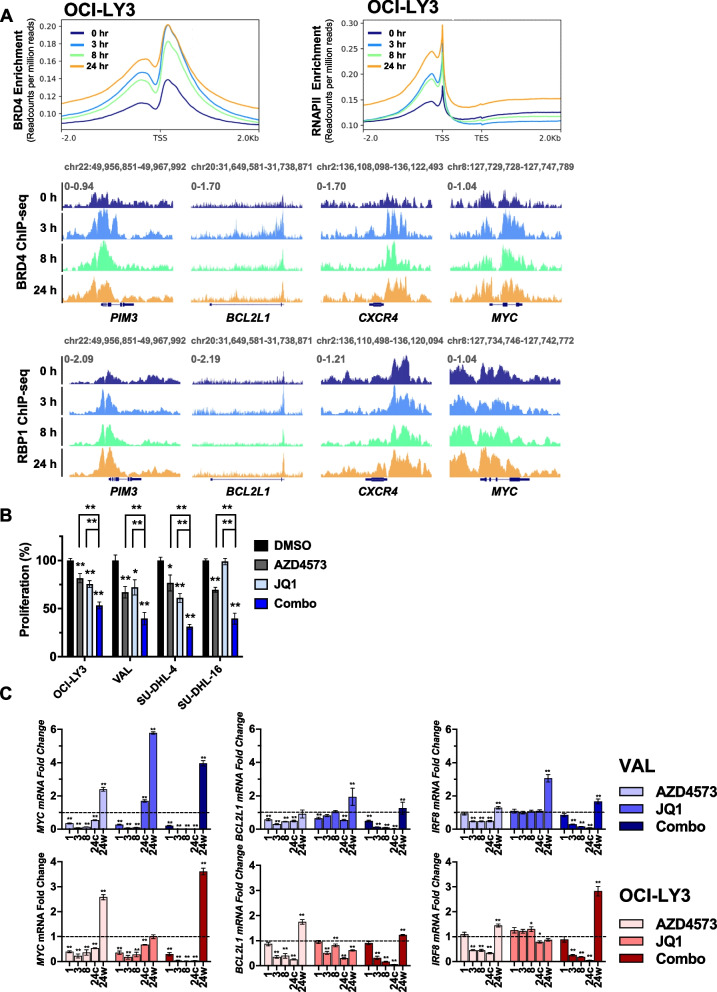
Diffuse large B-cell lymphoma (DLBCL) exhibits significant genetic heterogeneity which contributes to drug resistance, necessitating development of novel therapeutic approaches. Pharmacological inhibitors of cyclin-dependent kinases (CDK) demonstrated pre-clinical activity in DLBCL, however many stalled in clinical development. Here we show that AZD4573, a selective inhibitor of CDK9, restricted growth of DLBCL cells. CDK9 inhibition (CDK9i) resulted in rapid changes in the transcriptome and proteome, with downmodulation of multiple oncoproteins (eg, MYC, Mcl-1, JunB, PIM3) and deregulation of phosphoinotiside-3 kinase (PI3K) and senescence pathways. Following initial transcriptional repression due to RNAPII pausing, we observed transcriptional recovery of several oncogenes, including MYC and PIM3. ATAC-Seq and ChIP-Seq experiments revealed that CDK9i induced epigenetic remodeling with bi-directional changes in chromatin accessibility, suppressed promoter activation and led to sustained reprograming of the super-enhancer landscape. A CRISPR library screen suggested that SE-associated genes in the Mediator complex, as well as AKT1, confer resistance to CDK9i. Consistent with this, sgRNA-mediated knockout of MED12 sensitized cells to CDK9i. Informed by our mechanistic findings, we combined AZD4573 with either PIM kinase or PI3K inhibitors. Both combinations decreased proliferation and induced apoptosis in DLBCL and primary lymphoma cells in vitro as well as resulted in delayed tumor progression and extended survival of mice xenografted with DLBCL in vivo. Thus, CDK9i induces reprogramming of the epigenetic landscape, and super-enhancer driven recovery of select oncogenes may contribute to resistance to CDK9i. PIM and PI3K represent potential targets to circumvent resistance to CDK9i in the heterogeneous landscape of DLBCL.
Keywords: BRD4; CDK9; Mediator; PI3K; Super-enhancer.
© 2023. The Author(s).
Conflict of interest statement
A.V.D. has received consulting fees from Abbvie, AstraZeneca, Bayer Oncology, BeiGene, Bristol Meyers Squibb, Genentech, Incyte, Lilly Oncology, Morphposys, Nurix, Oncovalent, Pharmacyclics and TG Therapeutics and has ongoing research funding from Abbvie, AstraZeneca, Bayer Oncology, Bristol Meyers Squibb, Cyclacel, MEI Pharma, Nurix and Takeda Oncology.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
