

The EN-TEx resource of multi-tissue personal epigenomes & variant-impact models
- PMID: 37001506
- PMCID: PMC10074325
- DOI: 10.1016/j.cell.2023.02.018
The EN-TEx resource of multi-tissue personal epigenomes & variant-impact models
Abstract
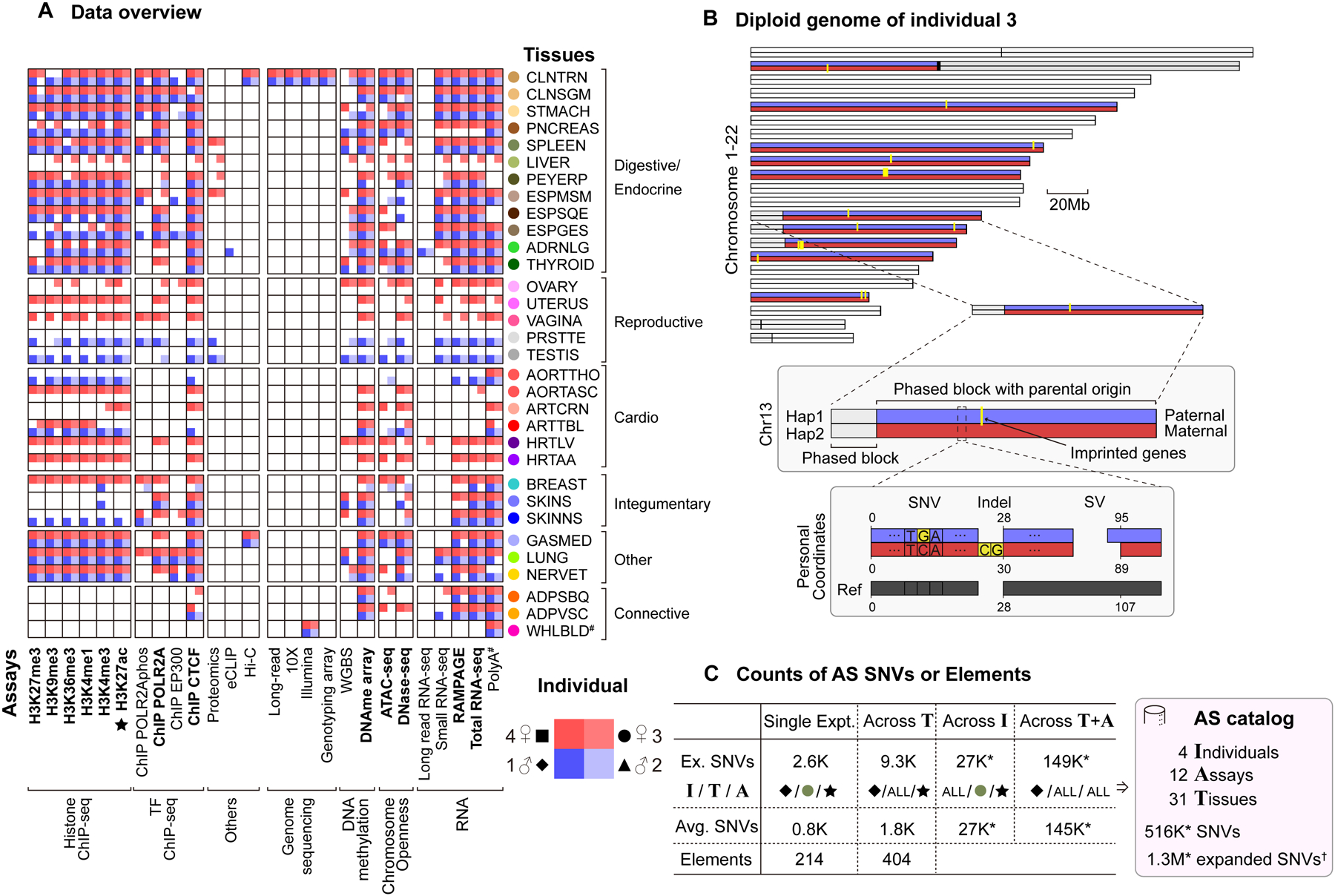
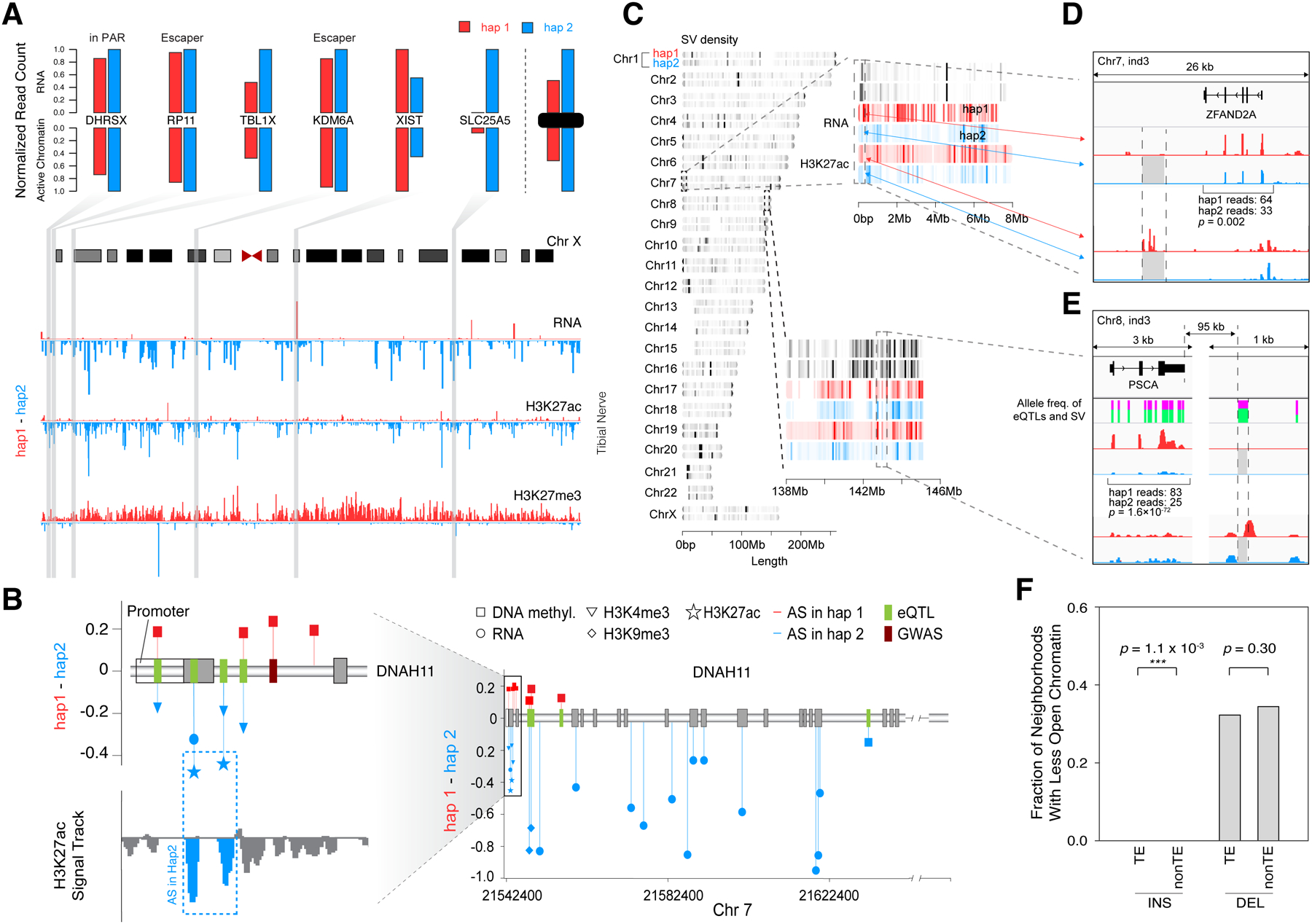
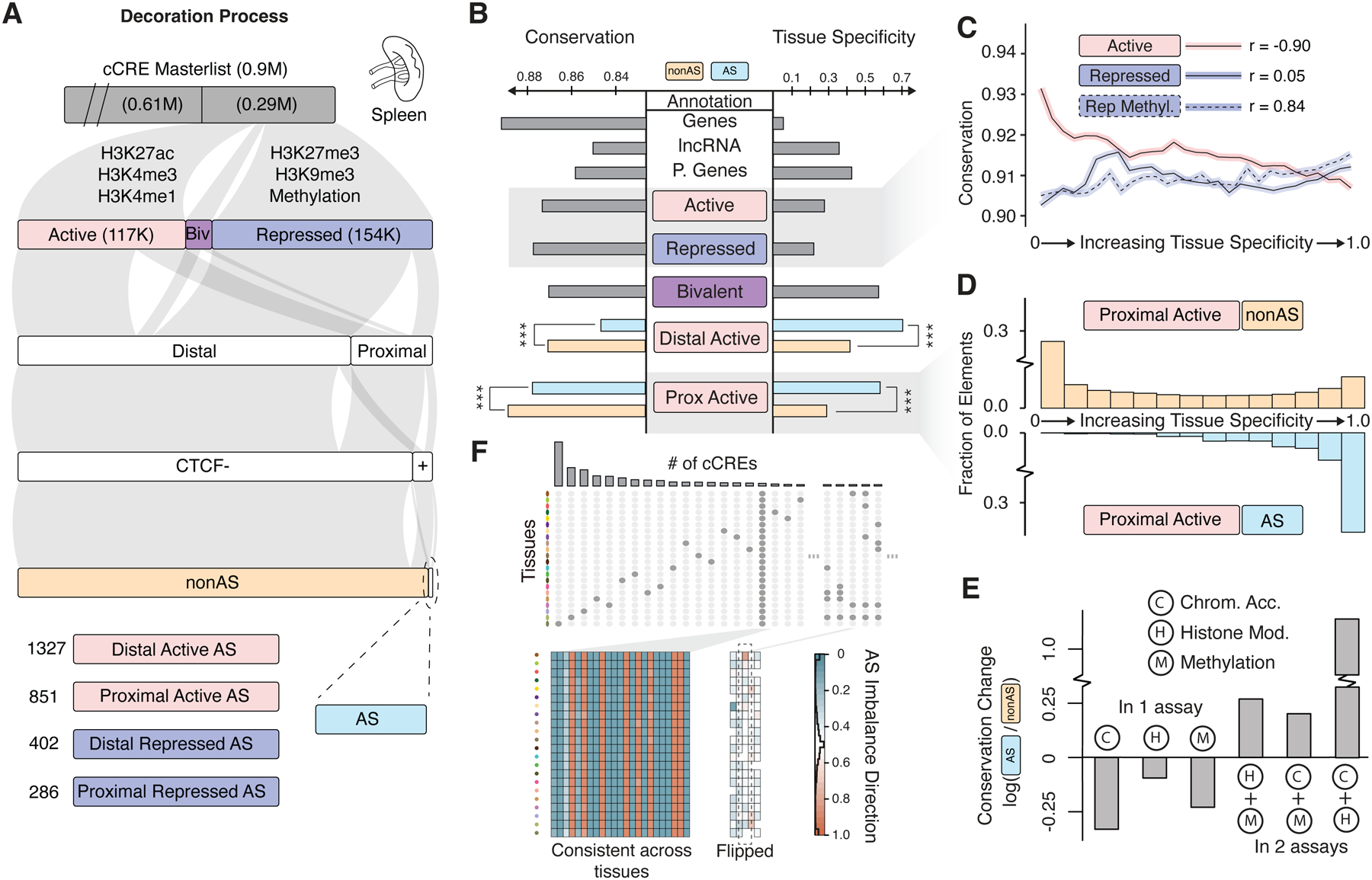
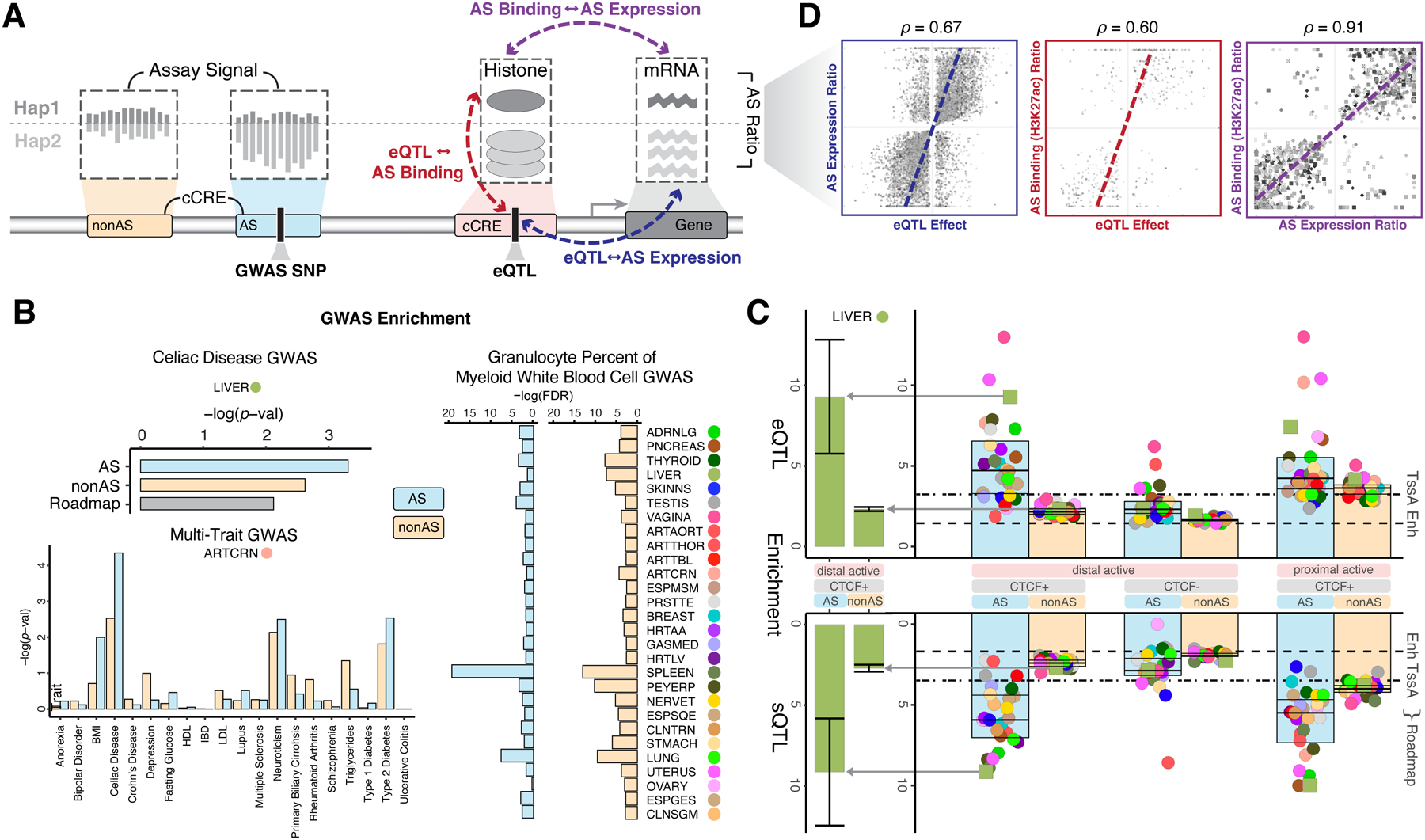
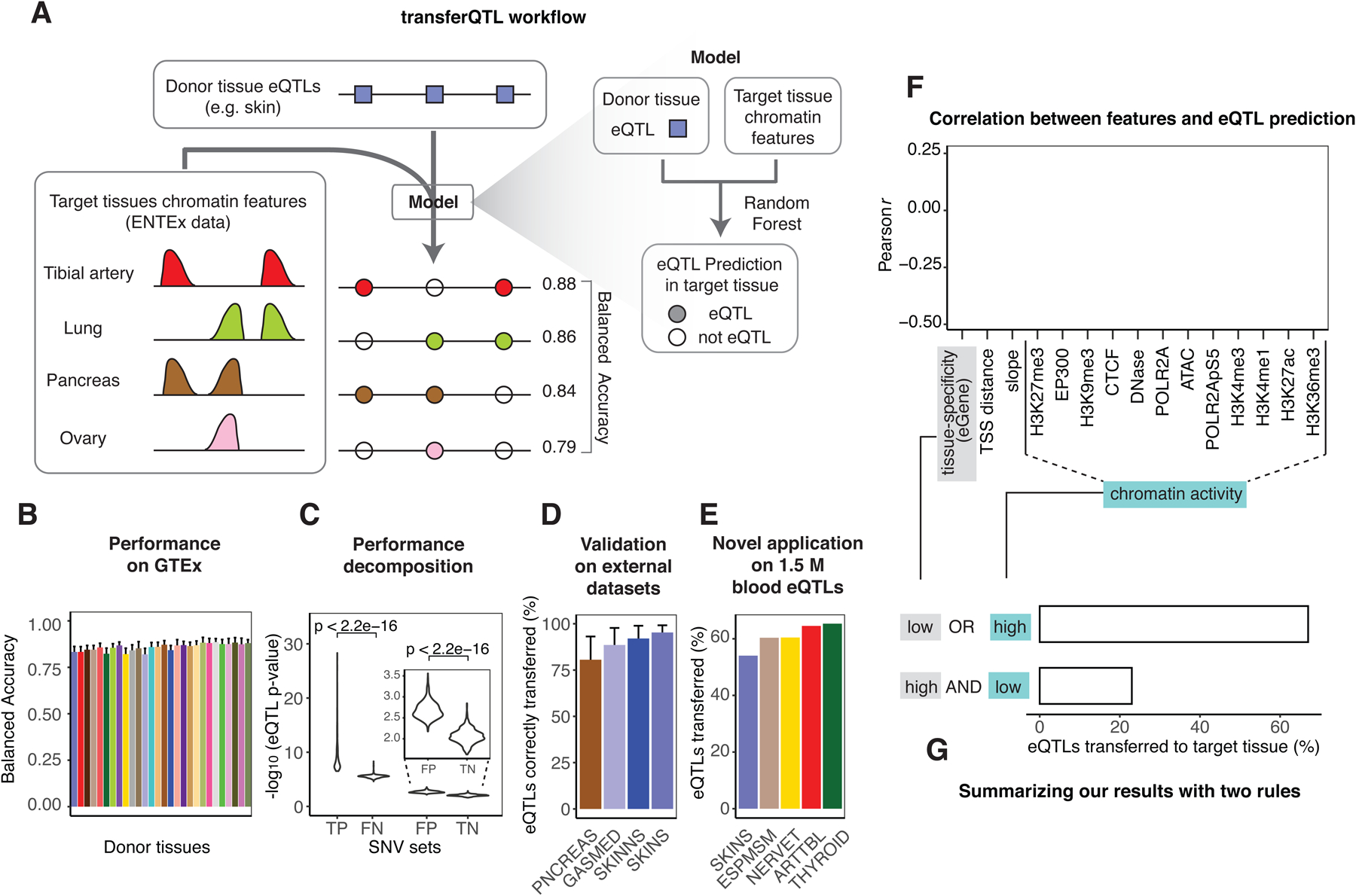
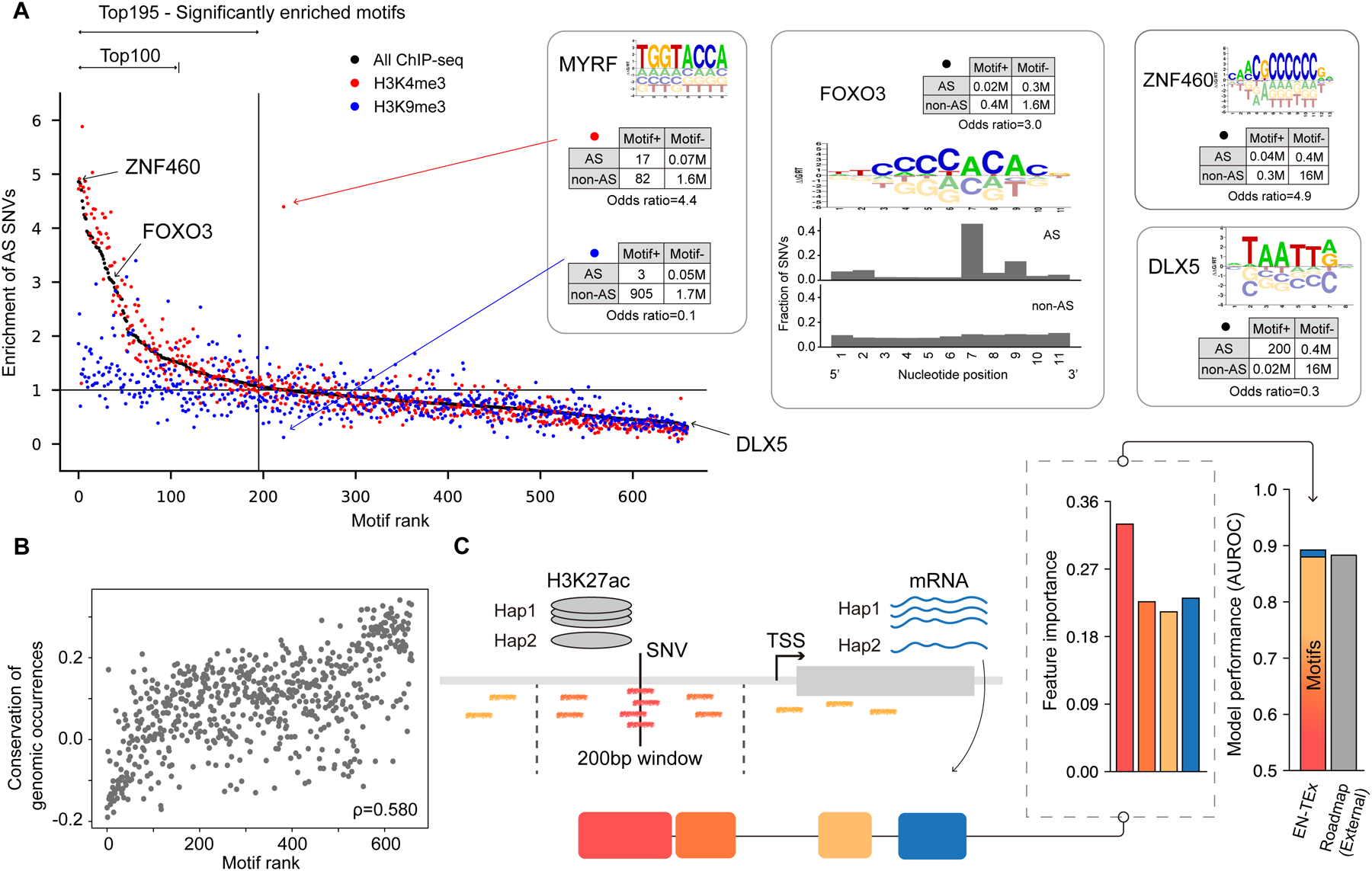
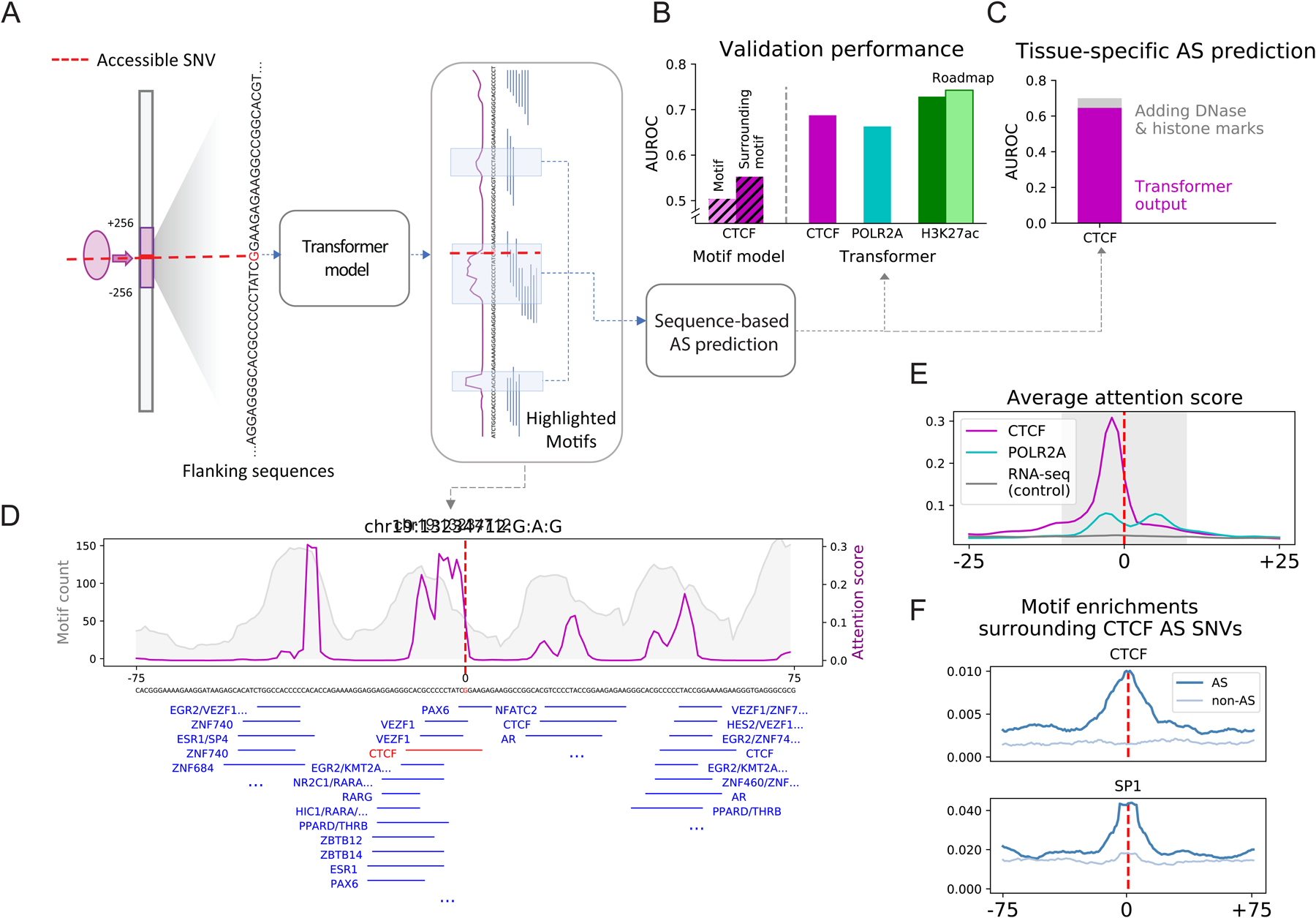
Understanding how genetic variants impact molecular phenotypes is a key goal of functional genomics, currently hindered by reliance on a single haploid reference genome. Here, we present the EN-TEx resource of 1,635 open-access datasets from four donors (∼30 tissues × ∼15 assays). The datasets are mapped to matched, diploid genomes with long-read phasing and structural variants, instantiating a catalog of >1 million allele-specific loci. These loci exhibit coordinated activity along haplotypes and are less conserved than corresponding, non-allele-specific ones. Surprisingly, a deep-learning transformer model can predict the allele-specific activity based only on local nucleotide-sequence context, highlighting the importance of transcription-factor-binding motifs particularly sensitive to variants. Furthermore, combining EN-TEx with existing genome annotations reveals strong associations between allele-specific and GWAS loci. It also enables models for transferring known eQTLs to difficult-to-profile tissues (e.g., from skin to heart). Overall, EN-TEx provides rich data and generalizable models for more accurate personal functional genomics.
Keywords: ENCODE; GTEx; allele-specific activity; eQTLs; functional epigenomes; functional genomics; genome annotations; personal genome; predictive models; structural variants; tissue specificity; transformer model.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests Z.W. co-founded and serves as a scientific advisor for Rgenta Inc. B.E.B. declares outside interests in Fulcrum Therapeutics, HiFiBio, Arsenal Biosciences, Cell Signaling Technologies, Chroma Medicine, and Design Pharmaceuticals. M.G. is on the advisory board for HypaHub, Inc. and Elysium Health.
Figures
Comment in
-
Epigenomes get personal.Nat Rev Genet. 2023 Jun;24(6):346. doi: 10.1038/s41576-023-00604-x. Nat Rev Genet. 2023. PMID: 37055612 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- U24 HG009397/HG/NHGRI NIH HHS/United States
- U54 HG006991/HG/NHGRI NIH HHS/United States
- U24 HG009649/HG/NHGRI NIH HHS/United States
- U54 HG007004/HG/NHGRI NIH HHS/United States
- R01 HG009318/HG/NHGRI NIH HHS/United States
- R01 MH113005/MH/NIMH NIH HHS/United States
- U01 CA253481/CA/NCI NIH HHS/United States
- U24 HG006620/HG/NHGRI NIH HHS/United States
- UM1 HG009390/HG/NHGRI NIH HHS/United States
- P30 CA045508/CA/NCI NIH HHS/United States
- U24 HG009446/HG/NHGRI NIH HHS/United States
- UM1 HG009442/HG/NHGRI NIH HHS/United States
- R01 LM012736/LM/NLM NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
