

Protein Structure Prediction: Challenges, Advances, and the Shift of Research Paradigms
- PMID: 37001856
- PMCID: PMC10928435
- DOI: 10.1016/j.gpb.2022.11.014
Protein Structure Prediction: Challenges, Advances, and the Shift of Research Paradigms
Abstract
Protein structure prediction is an interdisciplinary research topic that has attracted researchers from multiple fields, including biochemistry, medicine, physics, mathematics, and computer science. These researchers adopt various research paradigms to attack the same structure prediction problem: biochemists and physicists attempt to reveal the principles governing protein folding; mathematicians, especially statisticians, usually start from assuming a probability distribution of protein structures given a target sequence and then find the most likely structure, while computer scientists formulate protein structure prediction as an optimization problem - finding the structural conformation with the lowest energy or minimizing the difference between predicted structure and native structure. These research paradigms fall into the two statistical modeling cultures proposed by Leo Breiman, namely, data modeling and algorithmic modeling. Recently, we have also witnessed the great success of deep learning in protein structure prediction. In this review, we present a survey of the efforts for protein structure prediction. We compare the research paradigms adopted by researchers from different fields, with an emphasis on the shift of research paradigms in the era of deep learning. In short, the algorithmic modeling techniques, especially deep neural networks, have considerably improved the accuracy of protein structure prediction; however, theories interpreting the neural networks and knowledge on protein folding are still highly desired.
Keywords: Deep learning; Language model; Protein folding; Protein structure prediction; Transformer.
Copyright © 2023 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Fusong Ju and Jianwei Zhu are the current employees of Microsoft Corp. Qi Zhang is the current employee of Huawei Technologies Co., Ltd. All the other authors have declared no competing interests.
Figures

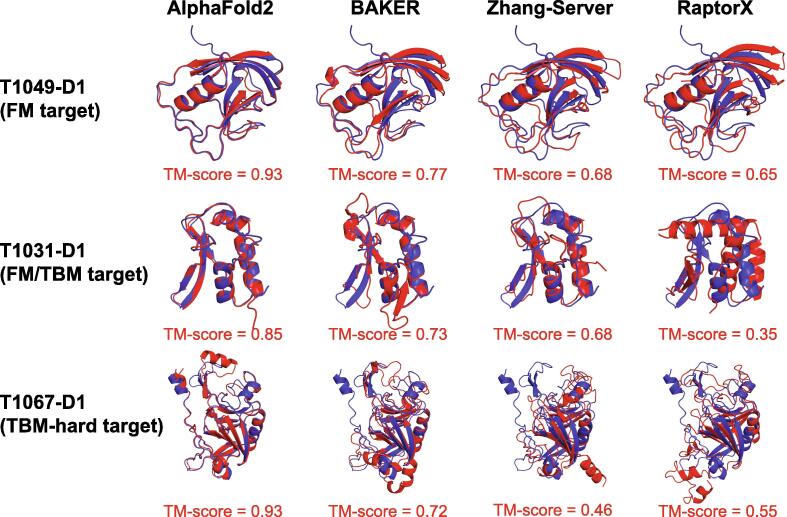
Similar articles
-
Protein tertiary structure modeling driven by deep learning and contact distance prediction in CASP13.Proteins. 2019 Dec;87(12):1165-1178. doi: 10.1002/prot.25697. Epub 2019 Apr 25. Proteins. 2019. PMID: 30985027 Free PMC article.
-
A novel model-based on FCM-LM algorithm for prediction of protein folding rate.J Bioinform Comput Biol. 2017 Aug;15(4):1750012. doi: 10.1142/S0219720017500123. Epub 2017 Apr 25. J Bioinform Comput Biol. 2017. PMID: 28513252
-
Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model.PLoS Comput Biol. 2017 Jan 5;13(1):e1005324. doi: 10.1371/journal.pcbi.1005324. eCollection 2017 Jan. PLoS Comput Biol. 2017. PMID: 28056090 Free PMC article.
-
Machine learning in protein structure prediction.Curr Opin Chem Biol. 2021 Dec;65:1-8. doi: 10.1016/j.cbpa.2021.04.005. Epub 2021 May 18. Curr Opin Chem Biol. 2021. PMID: 34015749 Review.
-
Deep Learning-Based Advances in Protein Structure Prediction.Int J Mol Sci. 2021 May 24;22(11):5553. doi: 10.3390/ijms22115553. Int J Mol Sci. 2021. PMID: 34074028 Free PMC article. Review.
Cited by
-
In silico approaches to study the human asparagine synthetase: An insight of the interaction between the enzyme active sites and its substrates.PLoS One. 2024 Aug 2;19(8):e0307448. doi: 10.1371/journal.pone.0307448. eCollection 2024. PLoS One. 2024. PMID: 39093903 Free PMC article.
-
In silico approaches supporting drug repurposing for Leishmaniasis: a scoping review.EXCLI J. 2024 Sep 3;23:1117-1169. doi: 10.17179/excli2024-7552. eCollection 2024. EXCLI J. 2024. PMID: 39421030 Free PMC article.
-
Characterization of soil-derived Bacillus subtilis metabolites against breast cancer: In vitro and in silico studies.Saudi Pharm J. 2025 Apr 17;33(1-2):3. doi: 10.1007/s44446-025-00006-6. Saudi Pharm J. 2025. PMID: 40397331 Free PMC article.
-
A comprehensive review of artificial intelligence for pharmacology research.Front Genet. 2024 Sep 3;15:1450529. doi: 10.3389/fgene.2024.1450529. eCollection 2024. Front Genet. 2024. PMID: 39290983 Free PMC article. Review.
-
An overview on olfaction in the biological, analytical, computational, and machine learning fields.Arch Pharm (Weinheim). 2025 Jan;358(1):e2400414. doi: 10.1002/ardp.202400414. Epub 2024 Oct 22. Arch Pharm (Weinheim). 2025. PMID: 39439128 Free PMC article. Review.
References
-
- Branden C., Tooze J. 2nd ed. Garland Science; New York: 1998. Introduction to protein structure.
-
- Finkelstein A.V., Ptitsyn O.B. 2nd ed. Elsevier; Amsterdam: 2016. Protein physics: a course of lectures.
-
- Kaur H., Garg A., Raghava G.P.S. PEPstr: a de novo method for tertiary structure prediction of small bioactive peptides. Protein Pept Lett. 2007;14:626–631. - PubMed
-
- Dill K.A., MacCallum J.L. The protein-folding problem, 50 years on. Science. 2012;338:1042–1046. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
