

Best practices for single-cell analysis across modalities
- PMID: 37002403
- PMCID: PMC10066026
- DOI: 10.1038/s41576-023-00586-w
Best practices for single-cell analysis across modalities
Abstract
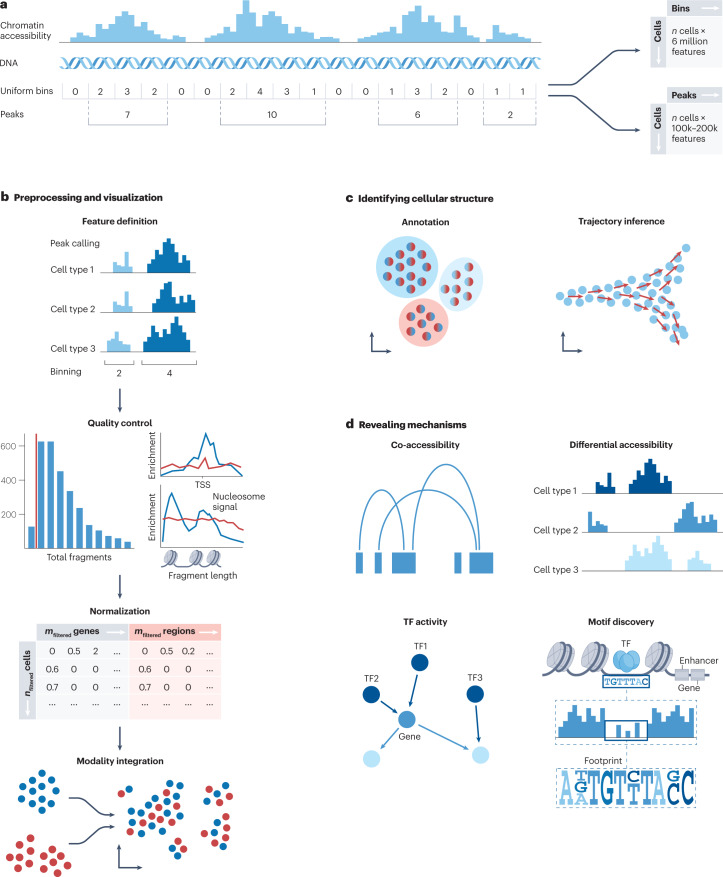
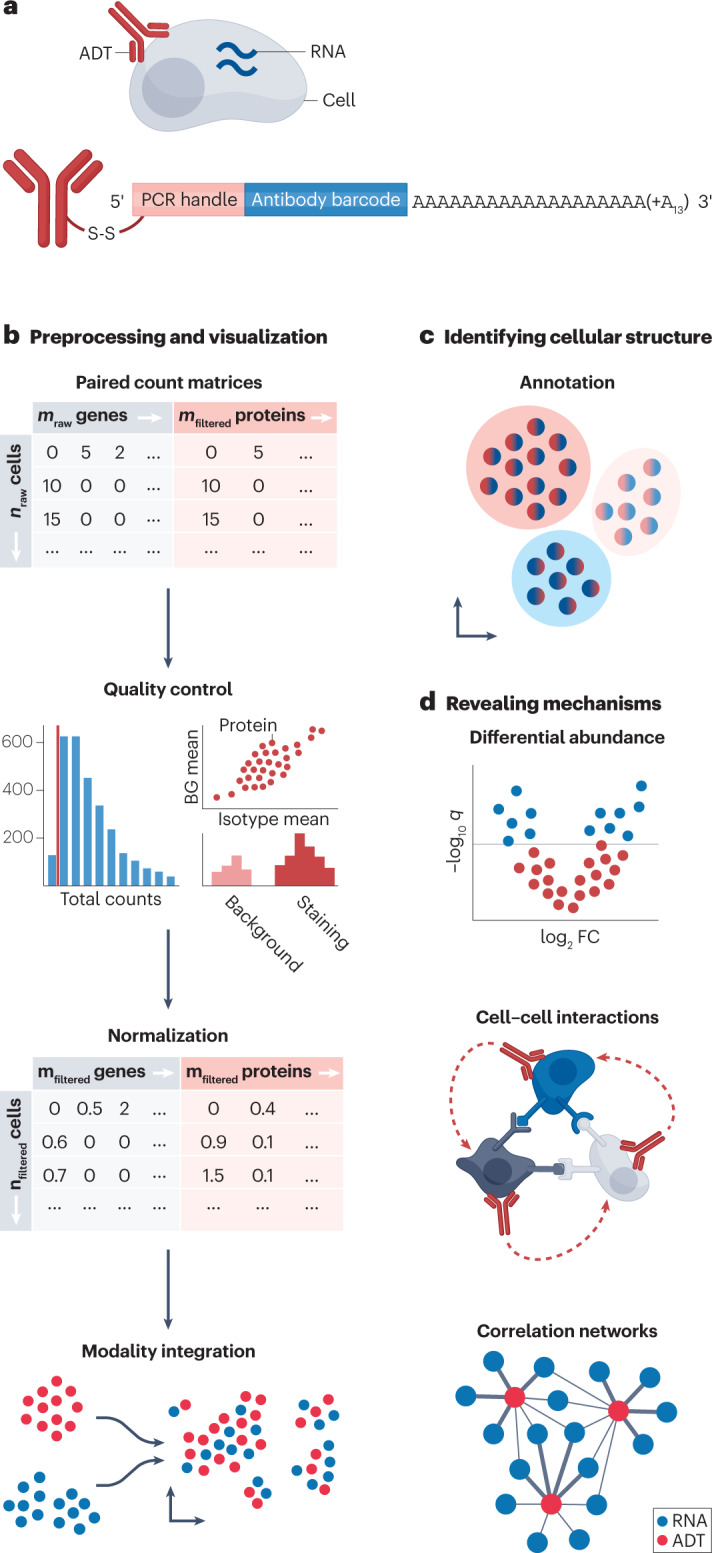
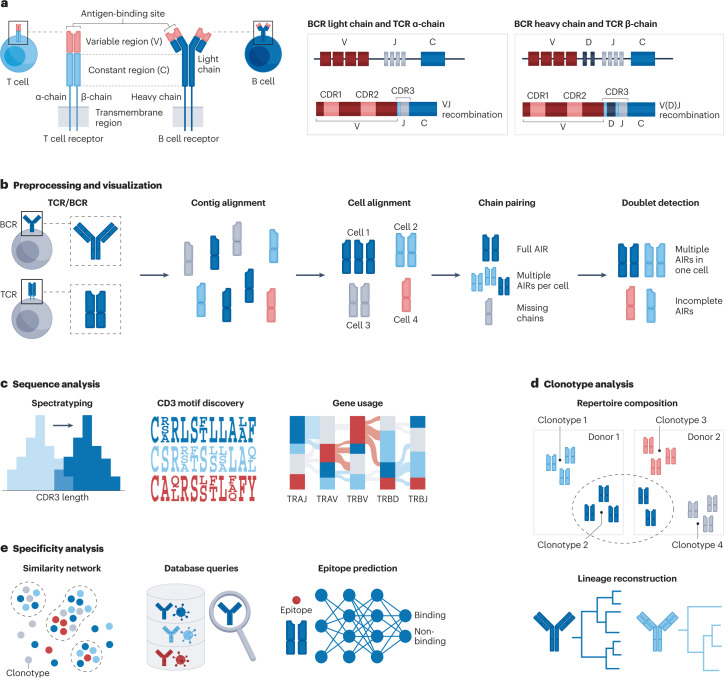
Recent advances in single-cell technologies have enabled high-throughput molecular profiling of cells across modalities and locations. Single-cell transcriptomics data can now be complemented by chromatin accessibility, surface protein expression, adaptive immune receptor repertoire profiling and spatial information. The increasing availability of single-cell data across modalities has motivated the development of novel computational methods to help analysts derive biological insights. As the field grows, it becomes increasingly difficult to navigate the vast landscape of tools and analysis steps. Here, we summarize independent benchmarking studies of unimodal and multimodal single-cell analysis across modalities to suggest comprehensive best-practice workflows for the most common analysis steps. Where independent benchmarks are not available, we review and contrast popular methods. Our article serves as an entry point for novices in the field of single-cell (multi-)omic analysis and guides advanced users to the most recent best practices.
© 2023. Springer Nature Limited.
Conflict of interest statement
Main author list: M.D.L. has received speaker’s honoraria from Pfizer and Janssen, and received consulting fees from Chan-Zuckerberg Initiative. F.J.T. consults for Immunai Inc., Singularity Bio B.V., CytoReason Ltd and Omniscope Ltd, and has ownership interest in Dermagnostix GmbH and Cellarity. M.G.J. consults for and has ownership interests in Vevo Therapeutics. L. Heumos has received speaker’s honorarium from Vesalius Therapeutics. Single-Cell Best Practices Consortium: M.G.J. consults for and has ownership interests in Vevo Therapeutics. R.P. is co-founder of Ocean Genomics, Inc. The other authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
