

Plant domestication shapes rhizosphere microbiome assembly and metabolic functions
- PMID: 37004105
- PMCID: PMC10064753
- DOI: 10.1186/s40168-023-01513-1
Plant domestication shapes rhizosphere microbiome assembly and metabolic functions
Abstract
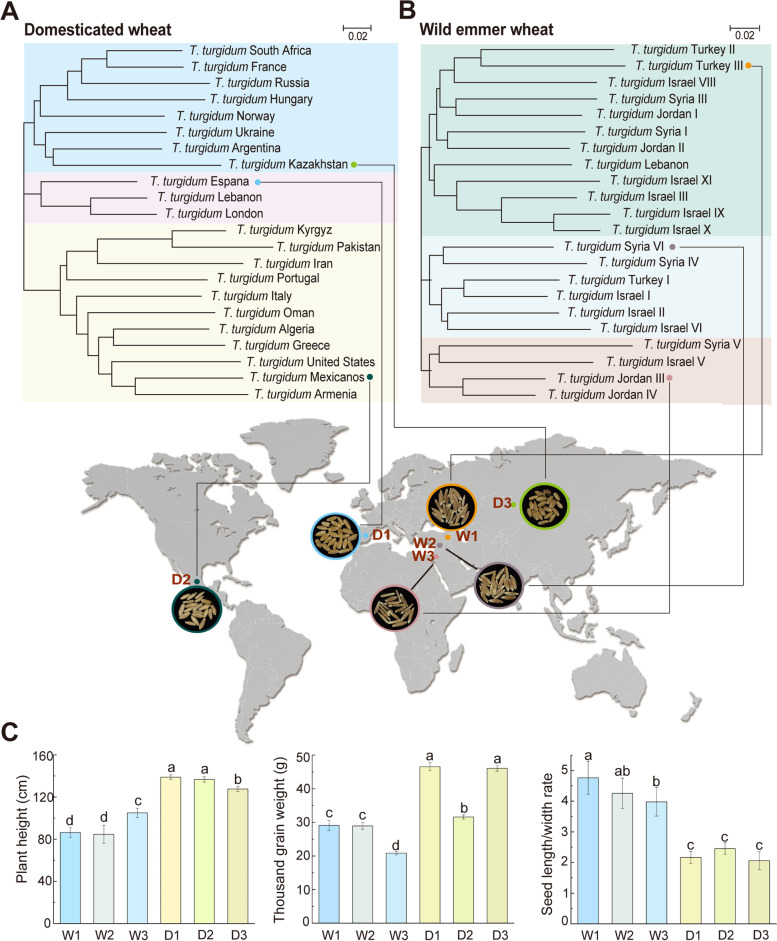
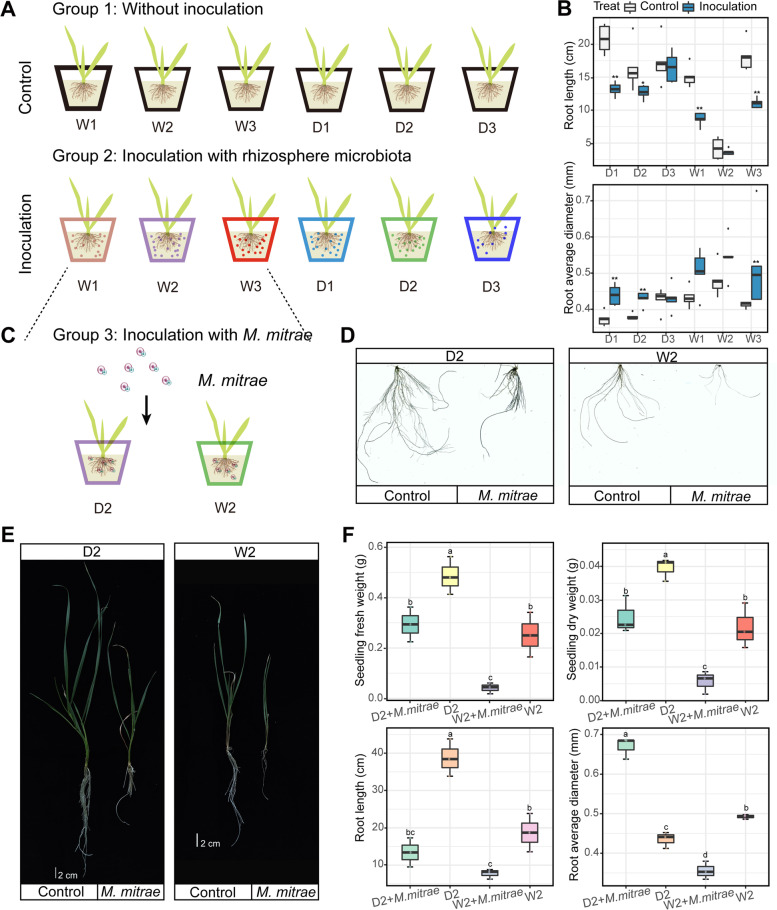
Background: The rhizosphere microbiome, which is shaped by host genotypes, root exudates, and plant domestication, is crucial for sustaining agricultural plant growth. Despite its importance, how plant domestication builds up specific rhizosphere microbiomes and metabolic functions, as well as the importance of these affected rhizobiomes and relevant root exudates in maintaining plant growth, is not well understood. Here, we firstly investigated the rhizosphere bacterial and fungal communities of domestication and wild accessions of tetraploid wheat using amplicon sequencing (16S and ITS) after 9 years of domestication process at the main production sites in China. We then explored the ecological roles of root exudation in shaping rhizosphere microbiome functions by integrating metagenomics and metabolic genomics approaches. Furthermore, we established evident linkages between root morphology traits and keystone taxa based on microbial culture and plant inoculation experiments.
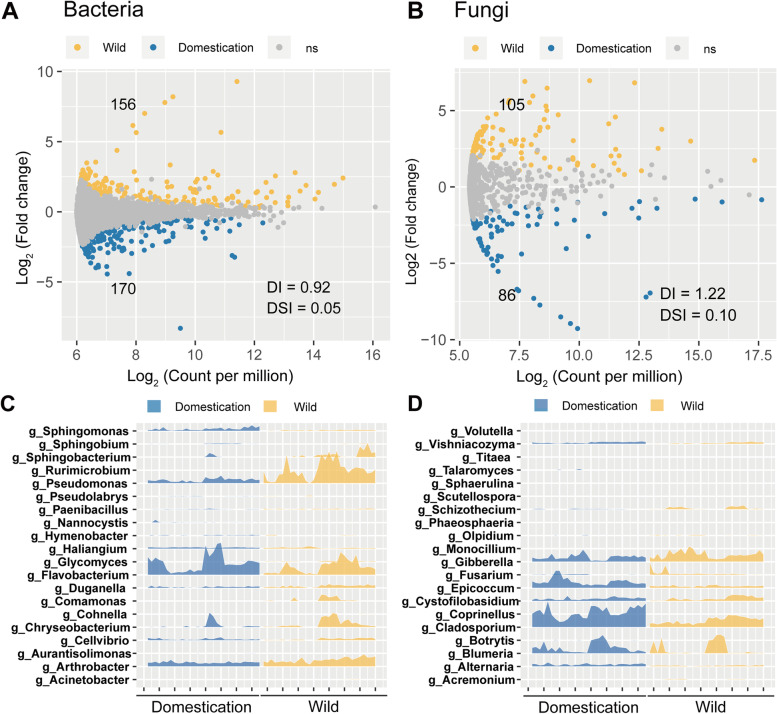
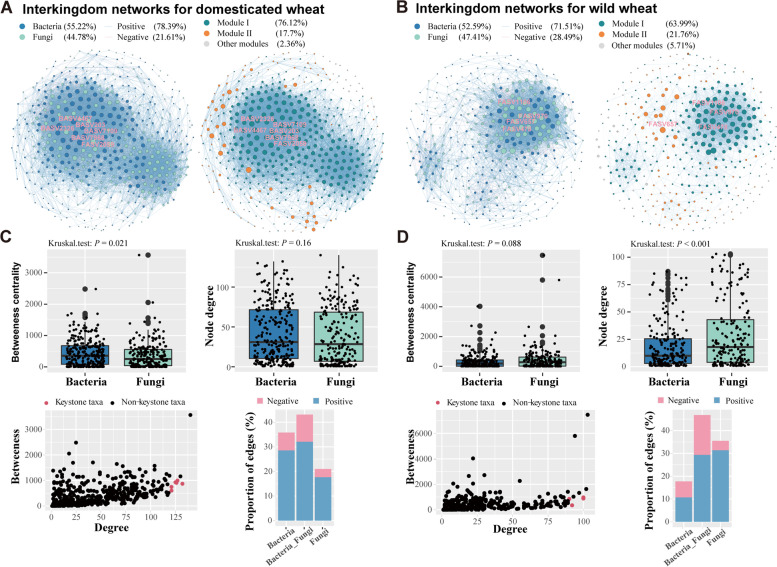
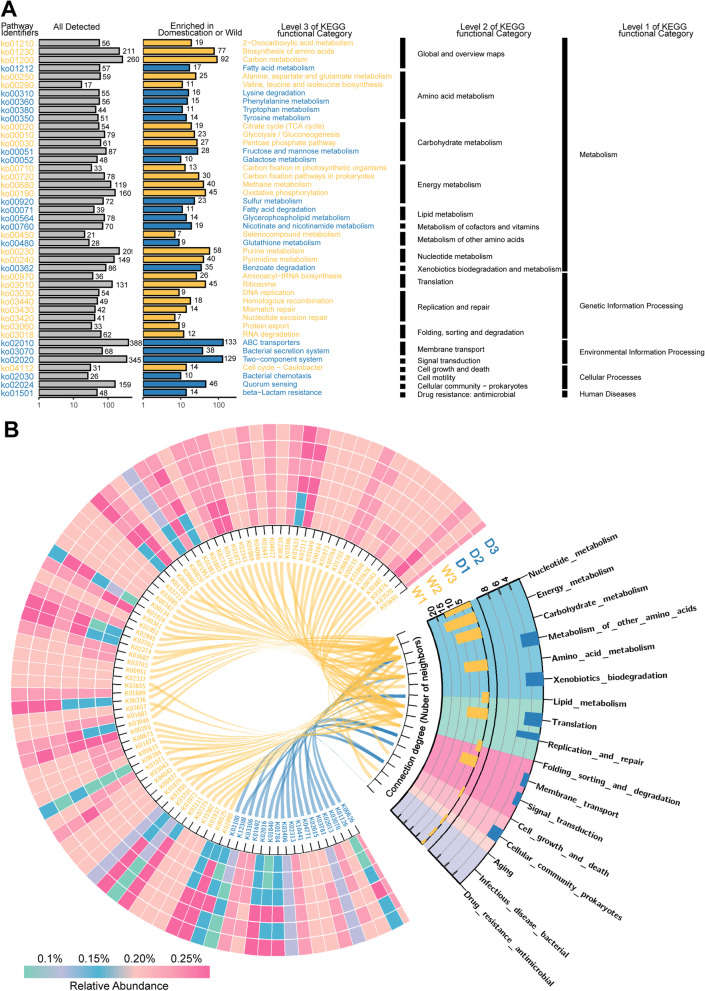
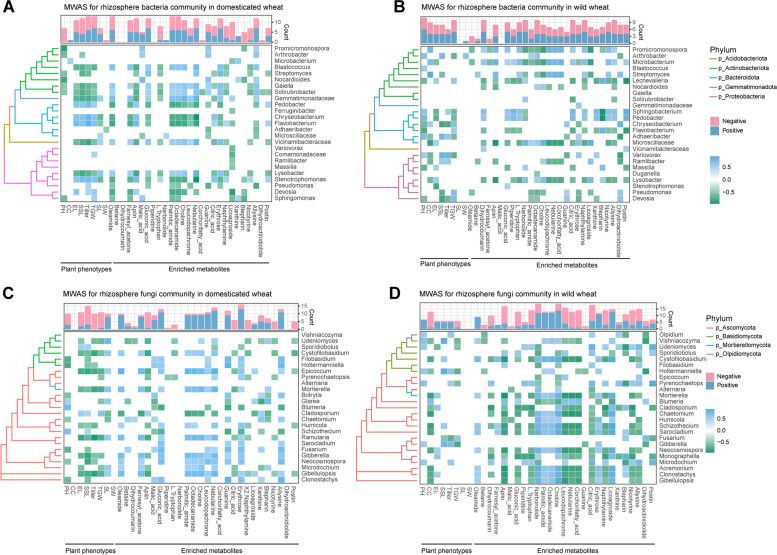
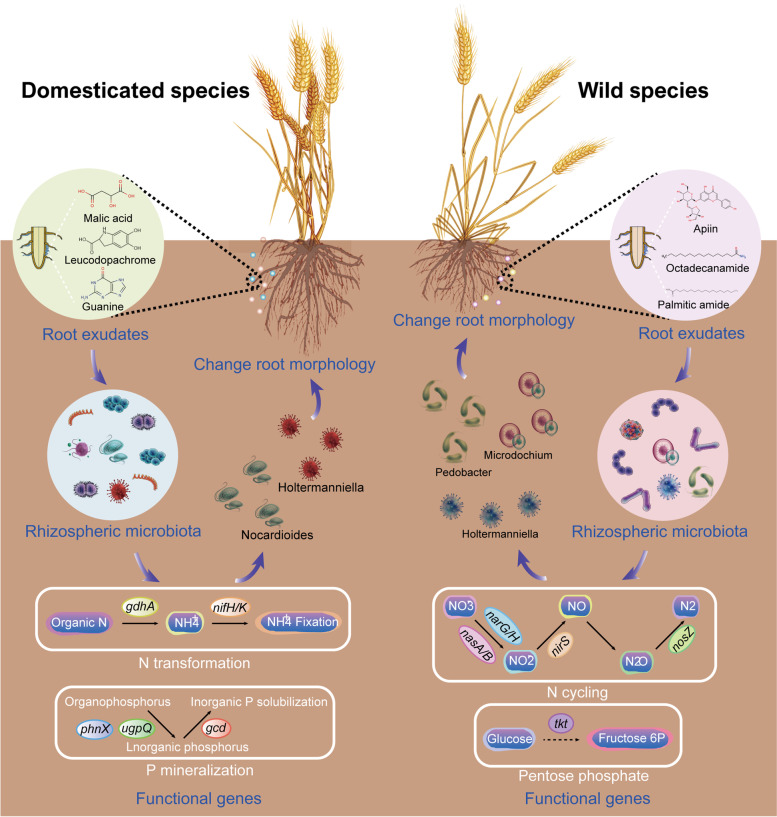
Results: Our results suggested that plant rhizosphere microbiomes were co-shaped by both host genotypes and domestication status. The wheat genomes contributed more variation in the microbial diversity and composition of rhizosphere bacterial communities than fungal communities, whereas plant domestication status exerted much stronger influences on the fungal communities. In terms of microbial interkingdom association networks, domestication destabilized microbial network and depleted the abundance of keystone fungal taxa. Moreover, we found that domestication shifted the rhizosphere microbiome from slow growing and fungi dominated to fast growing and bacteria dominated, thereby resulting in a shift from fungi-dominated membership with enrichment of carbon fixation genes to bacteria-dominated membership with enrichment of carbon degradation genes. Metagenomics analyses further indicated that wild cultivars of wheat possess higher microbial function diversity than domesticated cultivars. Notably, we found that wild cultivar is able to harness rhizosphere microorganism carrying N transformation (i.e., nitrification, denitrification) and P mineralization pathway, whereas rhizobiomes carrying inorganic N fixation, organic N ammonification, and inorganic P solubilization genes are recruited by the releasing of root exudates from domesticated wheat. More importantly, our metabolite-wide association study indicated that the contrasting functional roles of root exudates and the harnessed keystone microbial taxa with different nutrient acquisition strategies jointly determined the aboveground plant phenotypes. Furthermore, we observed that although domesticated and wild wheats recruited distinct microbial taxa and relevant functions, domestication-induced recruitment of keystone taxa led to a consistent growth regulation of root regardless of wheat domestication status.
Conclusions: Our results indicate that plant domestication profoundly influences rhizosphere microbiome assembly and metabolic functions and provide evidence that host plants are able to harness a differentiated ecological role of root-associated keystone microbiomes through the release of root exudates to sustain belowground multi-nutrient cycles and plant growth. These findings provide valuable insights into the mechanisms underlying plant-microbiome interactions and how to harness the rhizosphere microbiome for crop improvement in sustainable agriculture. Video Abstract.
Keywords: Microbial interaction network; Microbial metabolic functions; Plant domestication; Rhizosphere microbiomes; Root exudation; Wheat.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Plant development influences dynamic shifts in the root compartment microbiomes of wild and domesticated finger millet cultivars.BMC Microbiol. 2025 Apr 30;25(1):259. doi: 10.1186/s12866-025-03976-8. BMC Microbiol. 2025. PMID: 40301775 Free PMC article.
-
Host genotype-specific rhizosphere fungus enhances drought resistance in wheat.Microbiome. 2024 Mar 4;12(1):44. doi: 10.1186/s40168-024-01770-8. Microbiome. 2024. PMID: 38433268 Free PMC article.
-
Domestication-driven changes in plant traits associated with changes in the assembly of the rhizosphere microbiota in tetraploid wheat.Sci Rep. 2020 Jul 22;10(1):12234. doi: 10.1038/s41598-020-69175-9. Sci Rep. 2020. PMID: 32699344 Free PMC article.
-
Effects of Domestication on Plant-Microbiome Interactions.Plant Cell Physiol. 2022 Nov 22;63(11):1654-1666. doi: 10.1093/pcp/pcac108. Plant Cell Physiol. 2022. PMID: 35876043 Review.
-
Root architecture and the rhizosphere microbiome: Shaping sustainable agriculture.Plant Sci. 2025 Oct;359:112599. doi: 10.1016/j.plantsci.2025.112599. Epub 2025 Jun 5. Plant Sci. 2025. PMID: 40482721 Review.
Cited by
-
Geographic Location Affects the Bacterial Community Composition and Diversity More than Species Identity for Tropical Tree Species.Plants (Basel). 2024 Jun 5;13(11):1565. doi: 10.3390/plants13111565. Plants (Basel). 2024. PMID: 38891373 Free PMC article.
-
The impact of salt-tolerant plants on soil nutrients and microbial communities in soda saline-alkali lands of the Songnen plain.Front Microbiol. 2025 Jun 5;16:1592834. doi: 10.3389/fmicb.2025.1592834. eCollection 2025. Front Microbiol. 2025. PMID: 40539100 Free PMC article.
-
Alpine radish rhizosphere microbiome assembly and metabolic adaptation under PBAT/PLA humic acid biodegradable mulch films.Front Microbiol. 2025 Jul 28;16:1623052. doi: 10.3389/fmicb.2025.1623052. eCollection 2025. Front Microbiol. 2025. PMID: 40792268 Free PMC article.
-
Assessing the Divergent Soil Phosphorus Recovery Strategies in Domesticated and Wild Crops.Plants (Basel). 2025 Jul 25;14(15):2296. doi: 10.3390/plants14152296. Plants (Basel). 2025. PMID: 40805645 Free PMC article. Review.
-
Enhanced understanding of nitrogen fixing bacteria through DNA extraction with polyvinylidene fluoride membrane.Sci Rep. 2025 May 8;15(1):16079. doi: 10.1038/s41598-025-00173-5. Sci Rep. 2025. PMID: 40341174 Free PMC article.
References
-
- Archer E. rfPermute: estimate permutation p-values for random forest importance metrics. R package version. 2016. p. 2.1.6.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
