

Integrative network-based analysis on multiple Gene Expression Omnibus datasets identifies novel immune molecular markers implicated in non-alcoholic steatohepatitis
- PMID: 37008925
- PMCID: PMC10061151
- DOI: 10.3389/fendo.2023.1115890
Integrative network-based analysis on multiple Gene Expression Omnibus datasets identifies novel immune molecular markers implicated in non-alcoholic steatohepatitis
Abstract
Introduction: Non-alcoholic steatohepatitis (NASH), an advanced subtype of non-alcoholic fatty liver disease (NAFLD), has becoming the most important aetiology for end-stage liver disease, such as cirrhosis and hepatocellular carcinoma. This study were designed to explore novel genes associated with NASH.
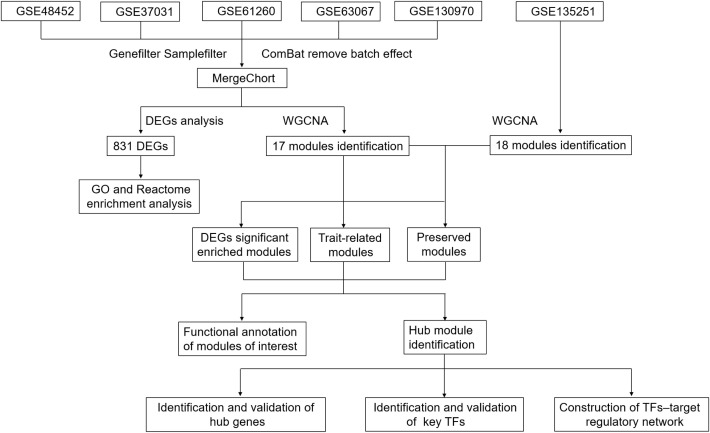
Methods: Here, five independent Gene Expression Omnibus (GEO) datasets were combined into a single cohort and analyzed using network biology approaches.
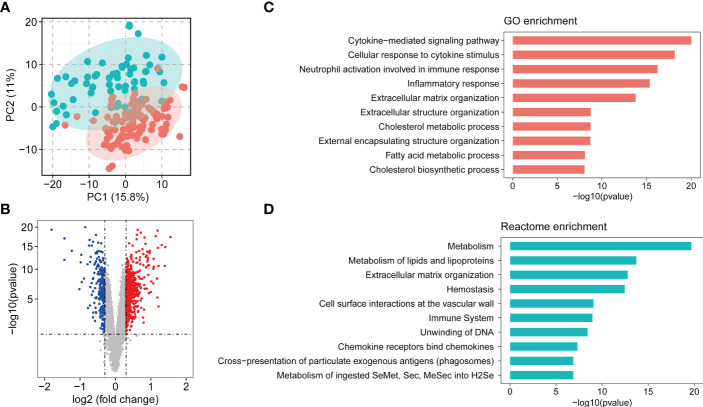
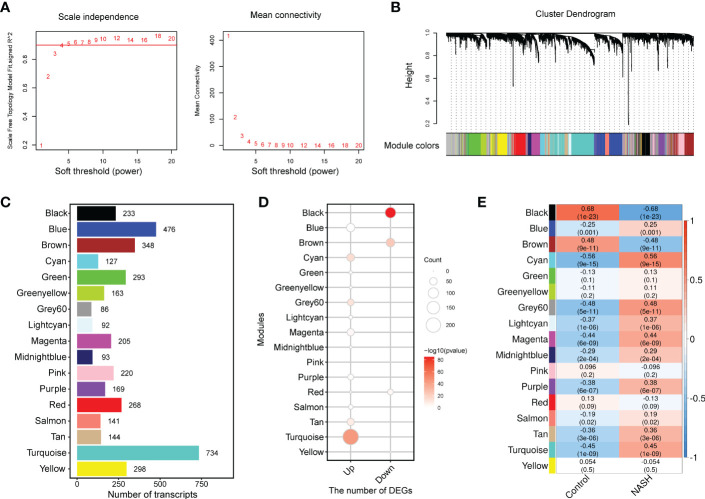
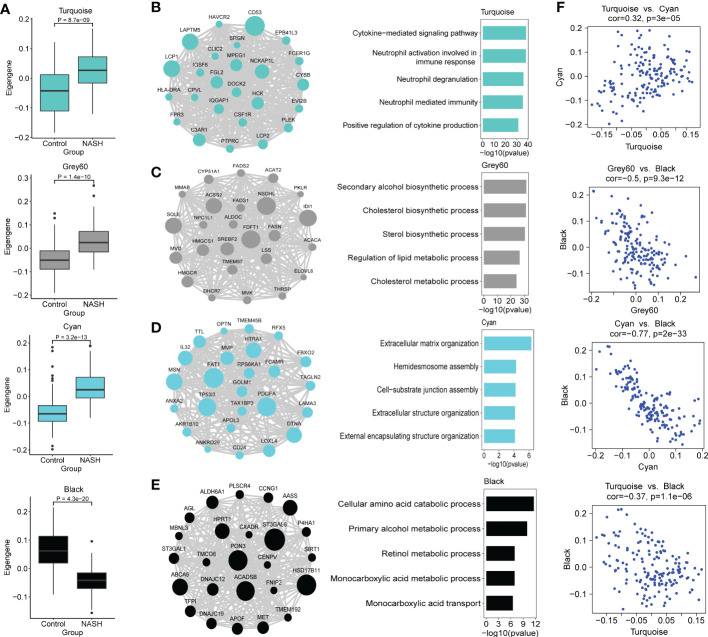
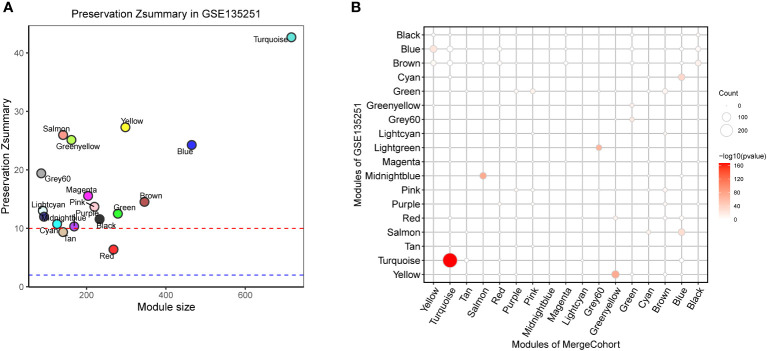
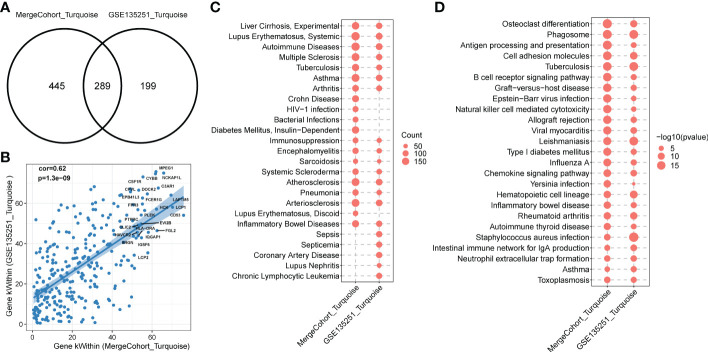
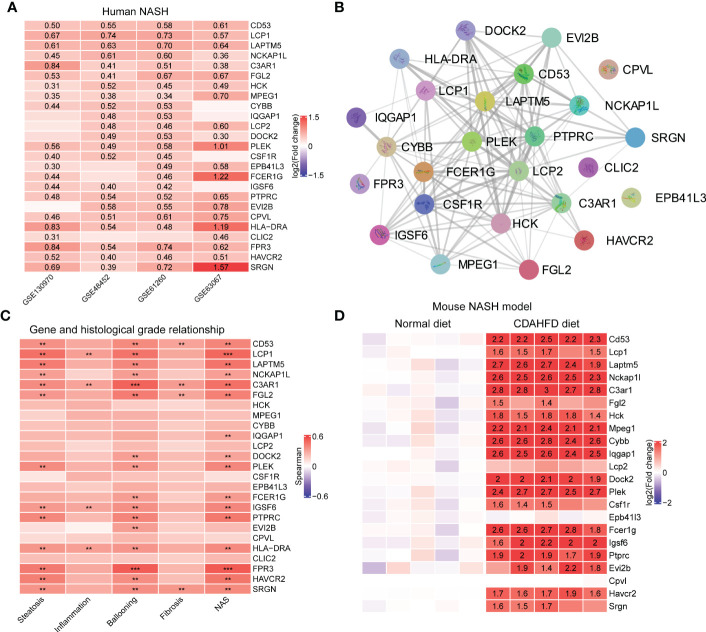
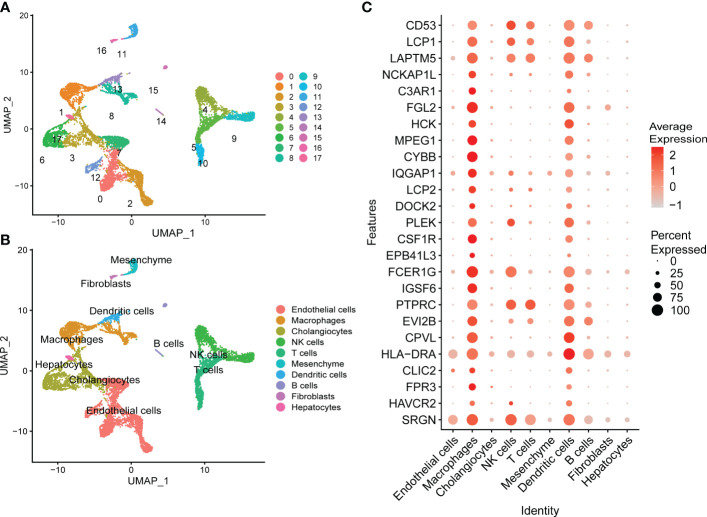
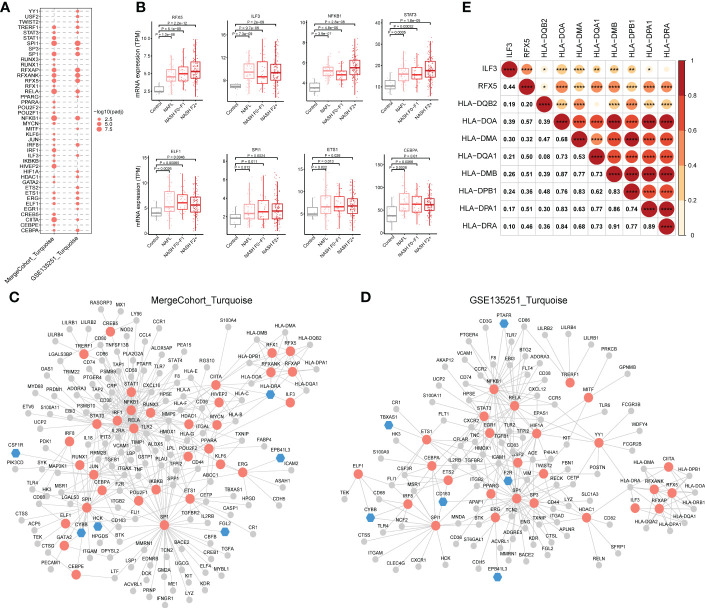
Results: 11 modules identified by weighted gene co-expression network analysis (WGCNA) showed significant association with the status of NASH. Further characterization of four gene modules of interest demonstrated that molecular pathology of NASH involves the upregulation of hub genes related to immune response, cholesterol and lipid metabolic process, extracellular matrix organization, and the downregulation of hub genes related to cellular amino acid catabolic, respectively. After DEGs enrichment analysis and module preservation analysis, the Turquoise module associated with immune response displayed a remarkably correlation with NASH status. Hub genes with high degree of connectivity in the module, including CD53, LCP1, LAPTM5, NCKAP1L, C3AR1, PLEK, FCER1G, HLA-DRA and SRGN were further verified in clinical samples and mouse model of NASH. Moreover, single-cell RNA-seq analysis showed that those key genes were expressed by distinct immune cells such as microphages, natural killer, dendritic, T and B cells. Finally, the potential transcription factors of Turquoise module were characterized, including NFKB1, STAT3, RFX5, ILF3, ELF1, SPI1, ETS1 and CEBPA, the expression of which increased with NASH progression.
Discussion: In conclusion, our integrative analysis will contribute to the understanding of NASH and may enable the development of potential biomarkers for NASH therapy.
Keywords: hub genes; immune response; non-alcoholic steatohepatitis; transcription factors; weighted gene co-expression network analysis.
Copyright © 2023 Zhang, Shen, Chen, Jiang, Yuan, Xie, Zhang and Xu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology (2011) 140:124–31. doi: 10.1053/j.gastro.2010.09.038 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous