

Targeting glutamine metabolism as a therapeutic strategy for cancer
- PMID: 37009798
- PMCID: PMC10167356
- DOI: 10.1038/s12276-023-00971-9
Targeting glutamine metabolism as a therapeutic strategy for cancer
Abstract
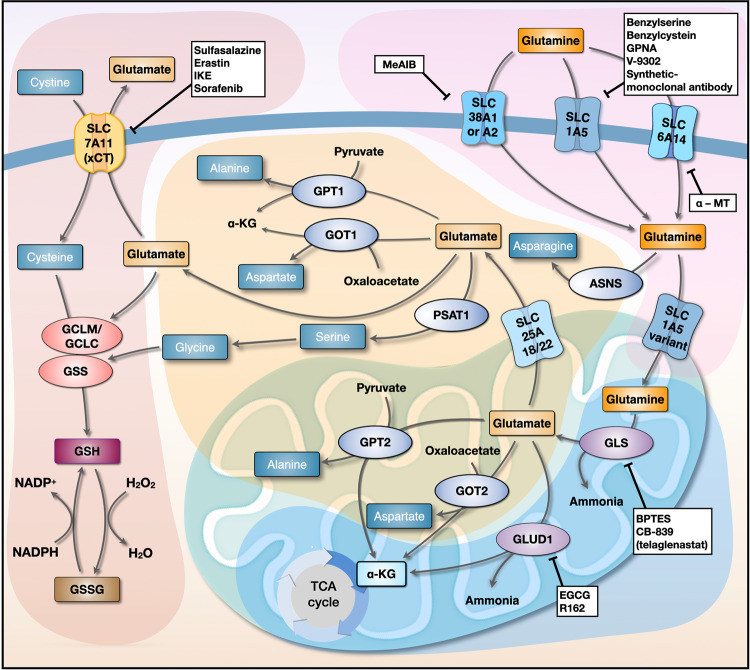
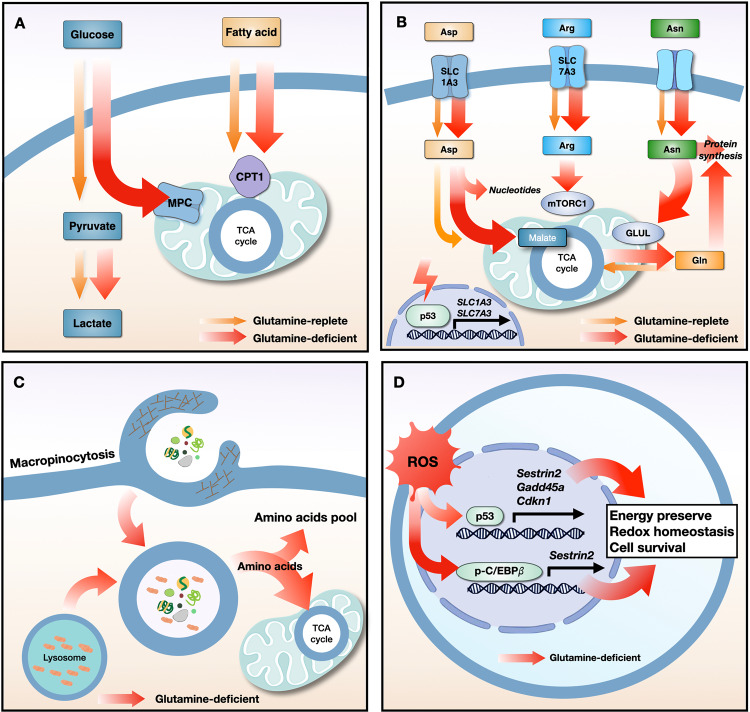
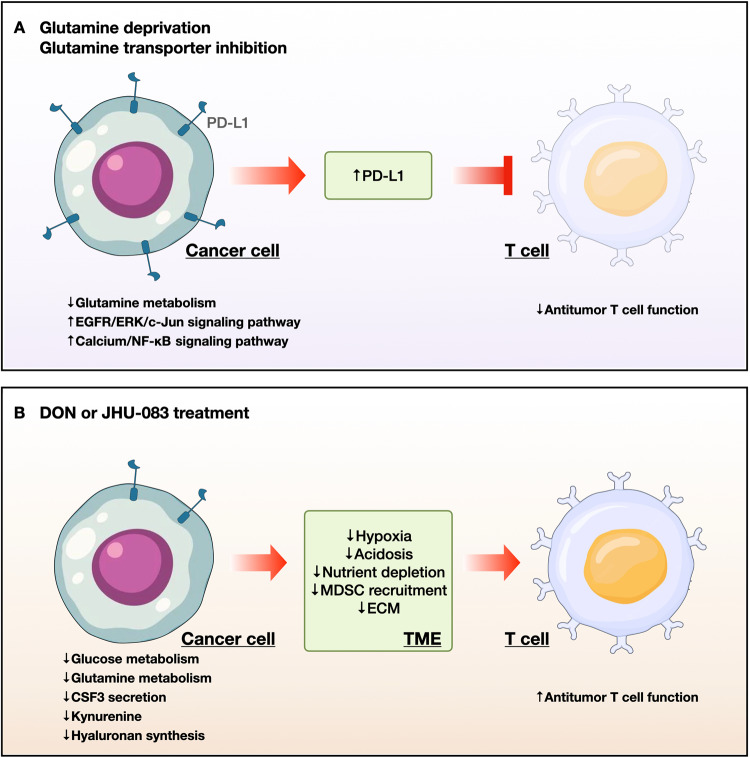
Proliferating cancer cells rely largely on glutamine for survival and proliferation. Glutamine serves as a carbon source for the synthesis of lipids and metabolites via the TCA cycle, as well as a source of nitrogen for amino acid and nucleotide synthesis. To date, many studies have explored the role of glutamine metabolism in cancer, thereby providing a scientific rationale for targeting glutamine metabolism for cancer treatment. In this review, we summarize the mechanism(s) involved at each step of glutamine metabolism, from glutamine transporters to redox homeostasis, and highlight areas that can be exploited for clinical cancer treatment. Furthermore, we discuss the mechanisms underlying cancer cell resistance to agents that target glutamine metabolism, as well as strategies for overcoming these mechanisms. Finally, we discuss the effects of glutamine blockade on the tumor microenvironment and explore strategies to maximize the utility of glutamine blockers as a cancer treatment.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
