

Comprehensive Functional Annotation of Metagenomes and Microbial Genomes Using a Deep Learning-Based Method
- PMID: 37010293
- PMCID: PMC10134832
- DOI: 10.1128/msystems.01178-22
Comprehensive Functional Annotation of Metagenomes and Microbial Genomes Using a Deep Learning-Based Method
Abstract
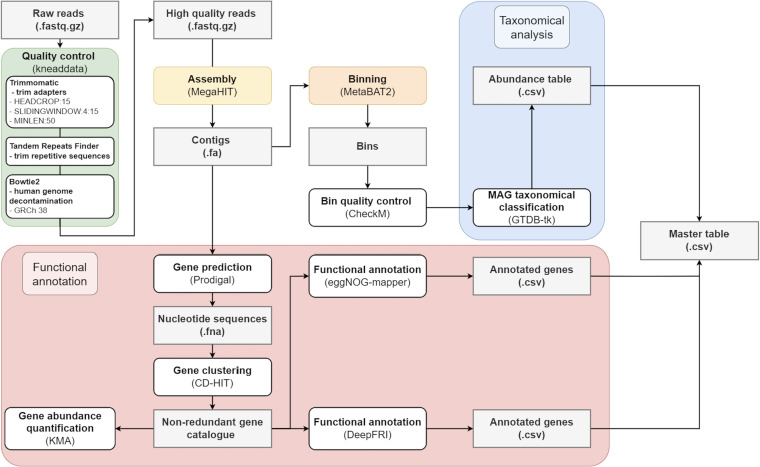
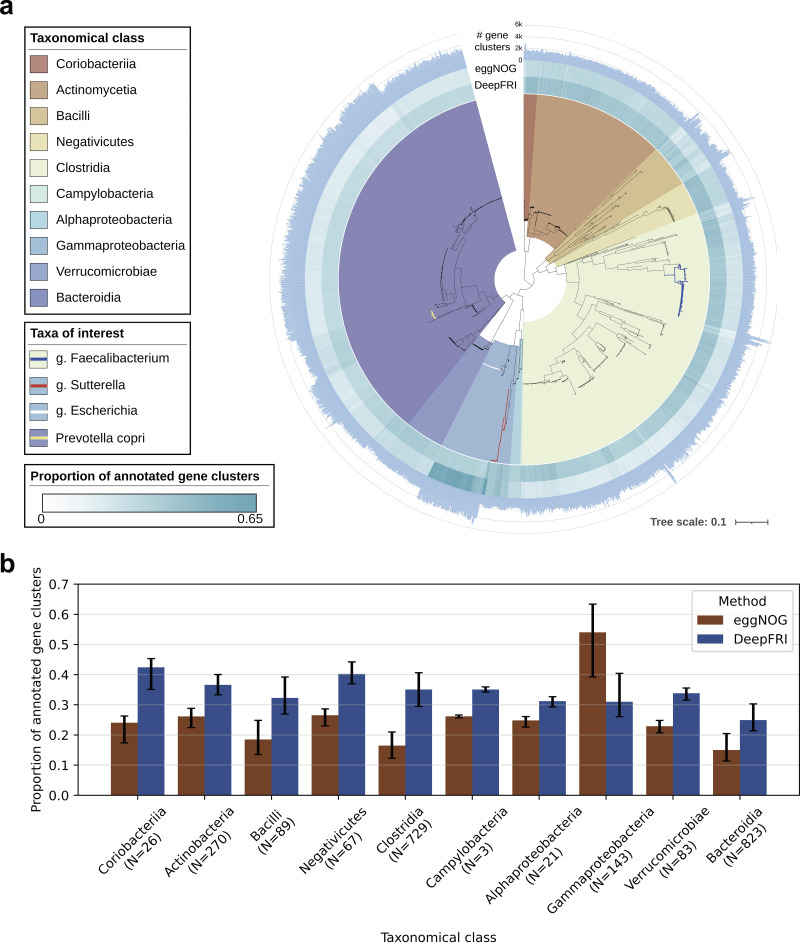
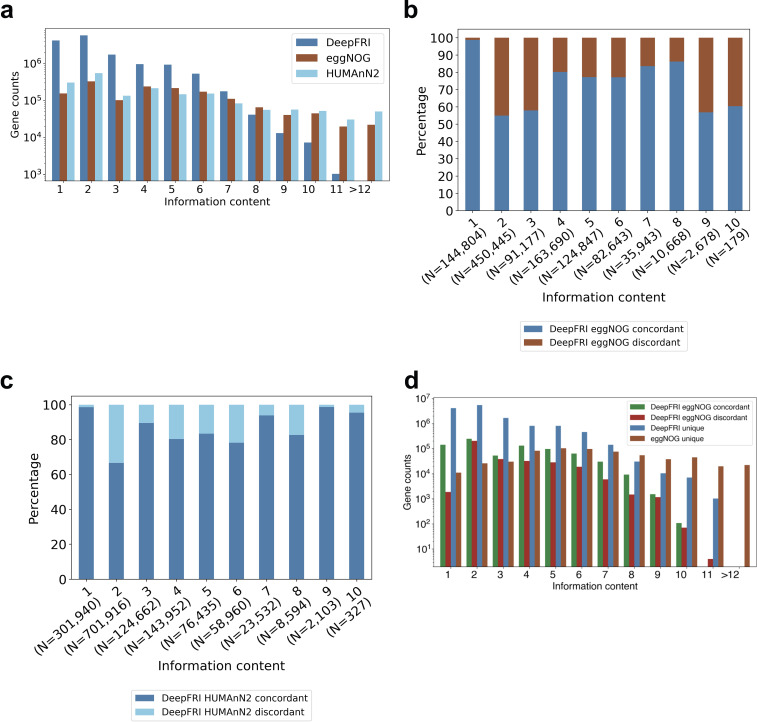
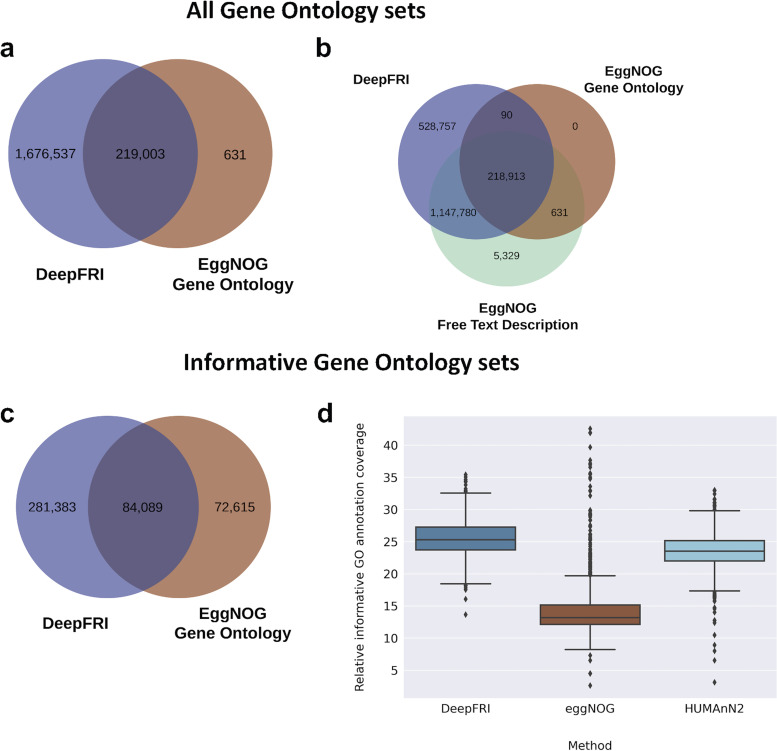
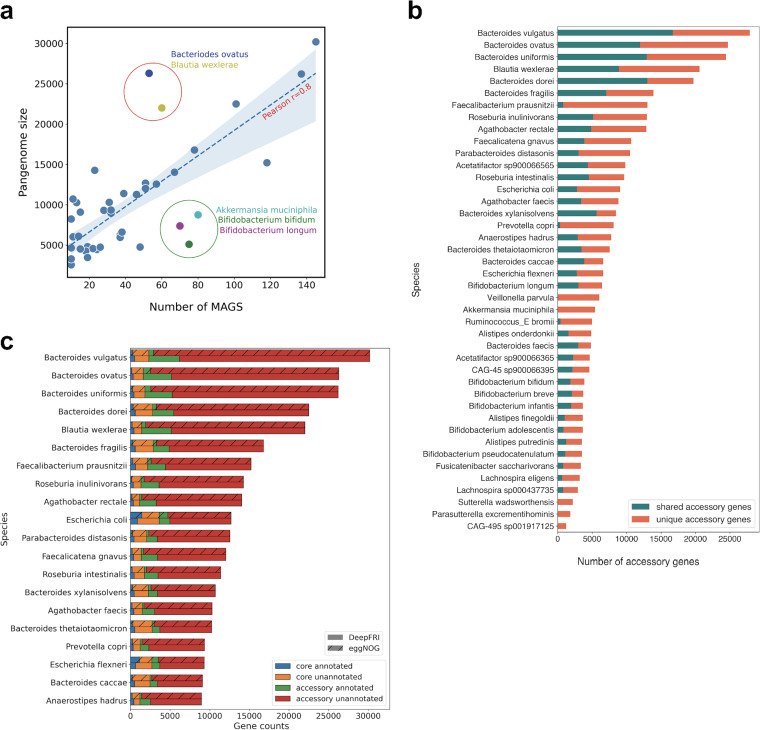
Comprehensive protein function annotation is essential for understanding microbiome-related disease mechanisms in the host organisms. However, a large portion of human gut microbial proteins lack functional annotation. Here, we have developed a new metagenome analysis workflow integrating de novo genome reconstruction, taxonomic profiling, and deep learning-based functional annotations from DeepFRI. This is the first approach to apply deep learning-based functional annotations in metagenomics. We validate DeepFRI functional annotations by comparing them to orthology-based annotations from eggNOG on a set of 1,070 infant metagenomes from the DIABIMMUNE cohort. Using this workflow, we generated a sequence catalogue of 1.9 million nonredundant microbial genes. The functional annotations revealed 70% concordance between Gene Ontology annotations predicted by DeepFRI and eggNOG. DeepFRI improved the annotation coverage, with 99% of the gene catalogue obtaining Gene Ontology molecular function annotations, although they are less specific than those from eggNOG. Additionally, we constructed pangenomes in a reference-free manner using high-quality metagenome-assembled genomes (MAGs) and analyzed the associated annotations. eggNOG annotated more genes on well-studied organisms, such as Escherichia coli, while DeepFRI was less sensitive to taxa. Further, we show that DeepFRI provides additional annotations in comparison to the previous DIABIMMUNE studies. This workflow will contribute to novel understanding of the functional signature of the human gut microbiome in health and disease as well as guiding future metagenomics studies. IMPORTANCE The past decade has seen advancement in high-throughput sequencing technologies resulting in rapid accumulation of genomic data from microbial communities. While this growth in sequence data and gene discovery is impressive, the majority of microbial gene functions remain uncharacterized. The coverage of functional information coming from either experimental sources or inferences is low. To solve these challenges, we have developed a new workflow to computationally assemble microbial genomes and annotate the genes using a deep learning-based model DeepFRI. This improved microbial gene annotation coverage to 1.9 million metagenome-assembled genes, representing 99% of the assembled genes, which is a significant improvement compared to 12% Gene Ontology term annotation coverage by commonly used orthology-based approaches. Importantly, the workflow supports pangenome reconstruction in a reference-free manner, allowing us to analyze the functional potential of individual bacterial species. We therefore propose this alternative approach combining deep-learning functional predictions with the commonly used orthology-based annotations as one that could help us uncover novel functions observed in metagenomic microbiome studies.
Keywords: deep learning; functional annotation; gene function; genome; metagenome; metagenome-assembled genomes; metagenomics; microbiome; orthology; pangenome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Accurate Annotation of Microbial Metagenomic Genes and Identification of Core Sets.Methods Mol Biol. 2021;2242:115-138. doi: 10.1007/978-1-0716-1099-2_8. Methods Mol Biol. 2021. PMID: 33961221
-
METABOLIC: high-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks.Microbiome. 2022 Feb 16;10(1):33. doi: 10.1186/s40168-021-01213-8. Microbiome. 2022. PMID: 35172890 Free PMC article.
-
Human reference gut microbiome catalog including newly assembled genomes from under-represented Asian metagenomes.Genome Med. 2021 Aug 27;13(1):134. doi: 10.1186/s13073-021-00950-7. Genome Med. 2021. PMID: 34446072 Free PMC article.
-
Genome-resolved metagenomics using environmental and clinical samples.Brief Bioinform. 2021 Sep 2;22(5):bbab030. doi: 10.1093/bib/bbab030. Brief Bioinform. 2021. PMID: 33758906 Free PMC article. Review.
-
An Experimental Approach to Genome Annotation: This report is based on a colloquium sponsored by the American Academy of Microbiology held July 19-20, 2004, in Washington, DC.Washington (DC): American Society for Microbiology; 2004. Washington (DC): American Society for Microbiology; 2004. PMID: 33001599 Free Books & Documents. Review.
Cited by
-
Integrative Approaches to Soybean Resilience, Productivity, and Utility: A Review of Genomics, Computational Modeling, and Economic Viability.Plants (Basel). 2025 Feb 21;14(5):671. doi: 10.3390/plants14050671. Plants (Basel). 2025. PMID: 40094561 Free PMC article. Review.
-
Analysis of metagenomic data.Nat Rev Methods Primers. 2025;5:5. doi: 10.1038/s43586-024-00376-6. Epub 2025 Jan 23. Nat Rev Methods Primers. 2025. PMID: 40688383 Free PMC article.
-
Exploring protein natural diversity in environmental microbiomes with DeepMetagenome.Cell Rep Methods. 2024 Nov 18;4(11):100896. doi: 10.1016/j.crmeth.2024.100896. Epub 2024 Nov 7. Cell Rep Methods. 2024. PMID: 39515333 Free PMC article.
-
Comprehensive analysis of orthologous genes reveals functional dynamics and energy metabolism in the rhizospheric microbiome of Moringa oleifera.Funct Integr Genomics. 2025 Apr 7;25(1):82. doi: 10.1007/s10142-025-01580-7. Funct Integr Genomics. 2025. PMID: 40195156 Free PMC article.
-
Bioinformatic approaches to blood and tissue microbiome analyses: challenges and perspectives.Brief Bioinform. 2025 Mar 4;26(2):bbaf176. doi: 10.1093/bib/bbaf176. Brief Bioinform. 2025. PMID: 40269515 Free PMC article. Review.
References
-
- Li J, Wang J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, Juncker AS, Manichanh C, Chen B, Zhang W, Levenez F, Wang J, Xu X, Xiao L, Liang S, Zhang D, Zhang Z, Chen W, Zhao H, Al-Aama JY, Edris S, Yang H, Wang J, Hansen T, Nielsen HB, Brunak S, Kristiansen K, Guarner F, Pedersen O, Doré J, Ehrlich SD, Bork P. 2014. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 32:834–841. doi:10.1038/nbt.2942. - DOI - PubMed
-
- Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, Collado MC, Rice BL, DuLong C, Morgan XC, Golden CD, Quince C, Huttenhower C, Segata N. 2019. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176:649–662.E20. doi:10.1016/j.cell.2019.01.001. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
