

Prioritized mass spectrometry increases the depth, sensitivity and data completeness of single-cell proteomics
- PMID: 37012480
- PMCID: PMC10172113
- DOI: 10.1038/s41592-023-01830-1
Prioritized mass spectrometry increases the depth, sensitivity and data completeness of single-cell proteomics
Abstract
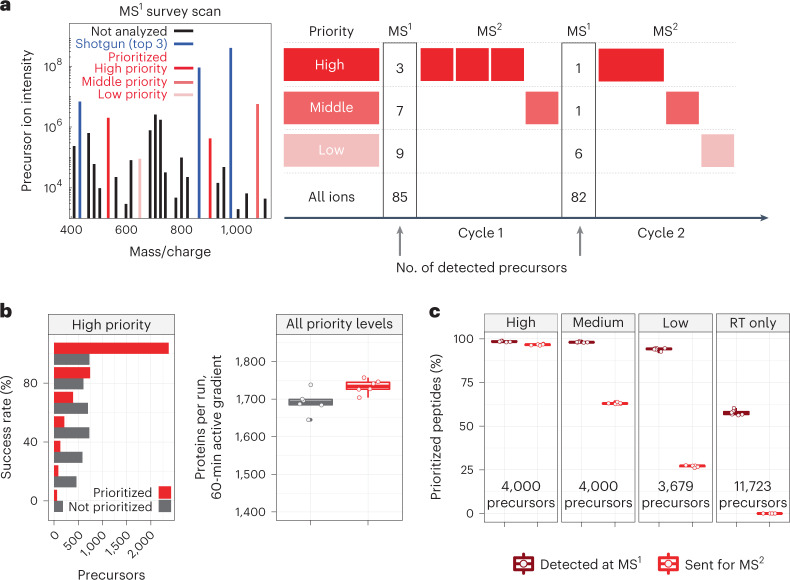
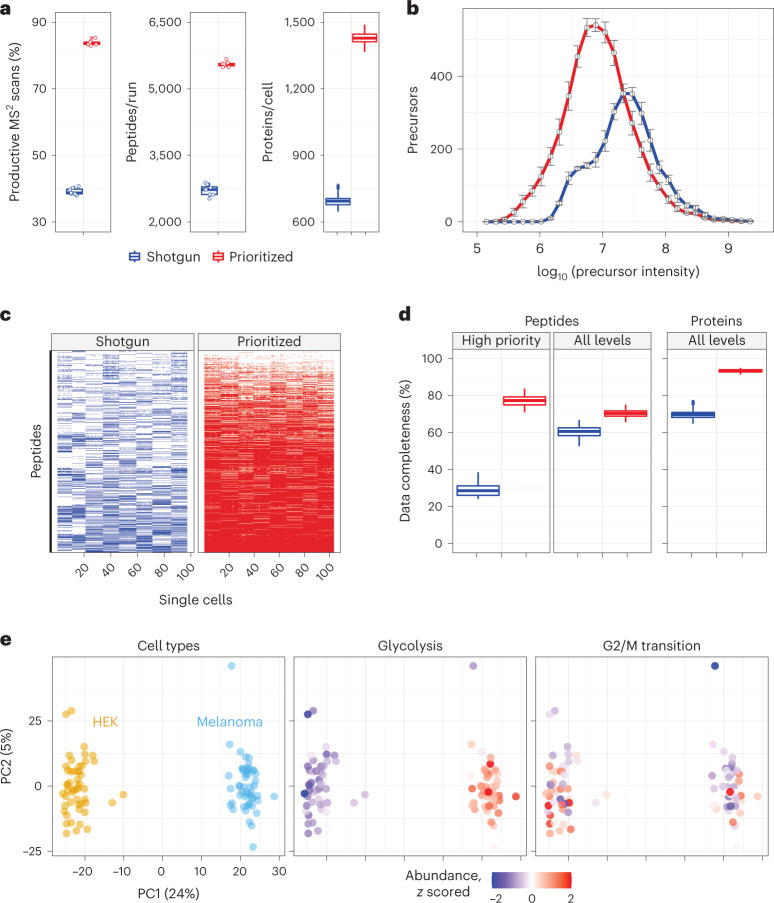
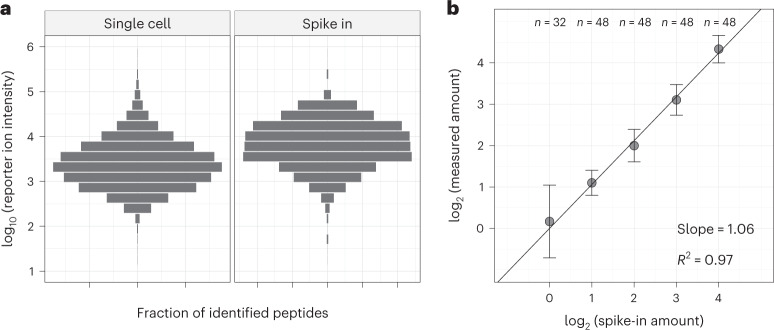
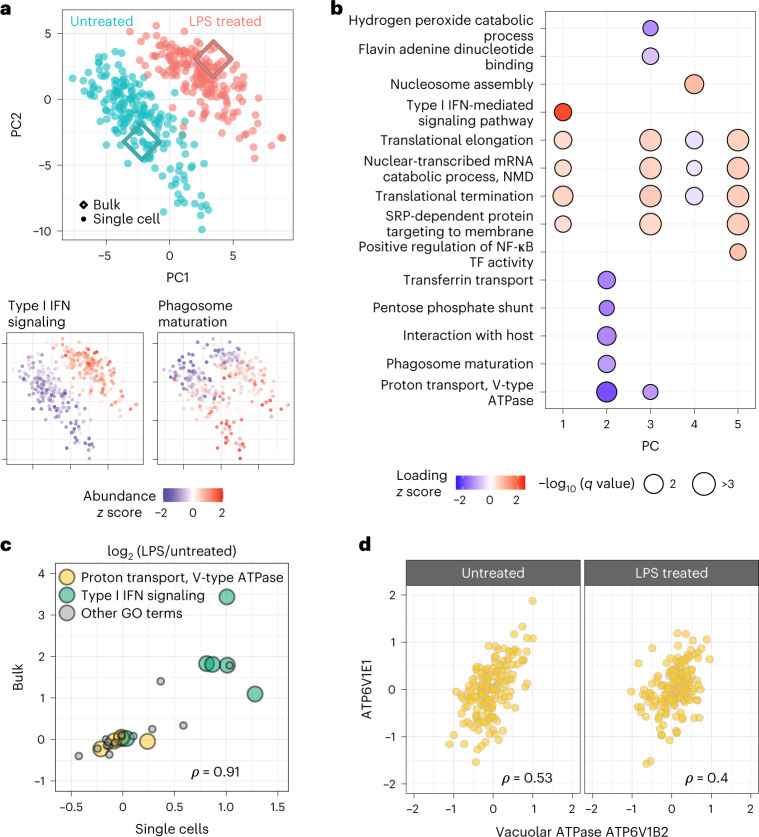
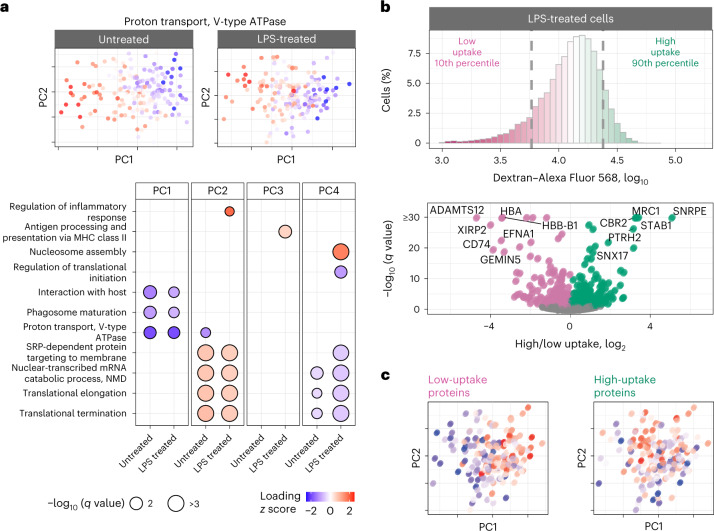
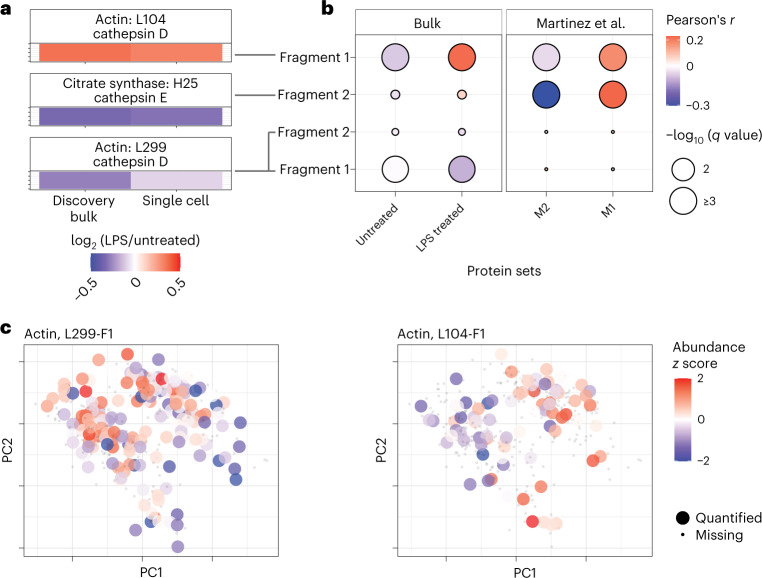
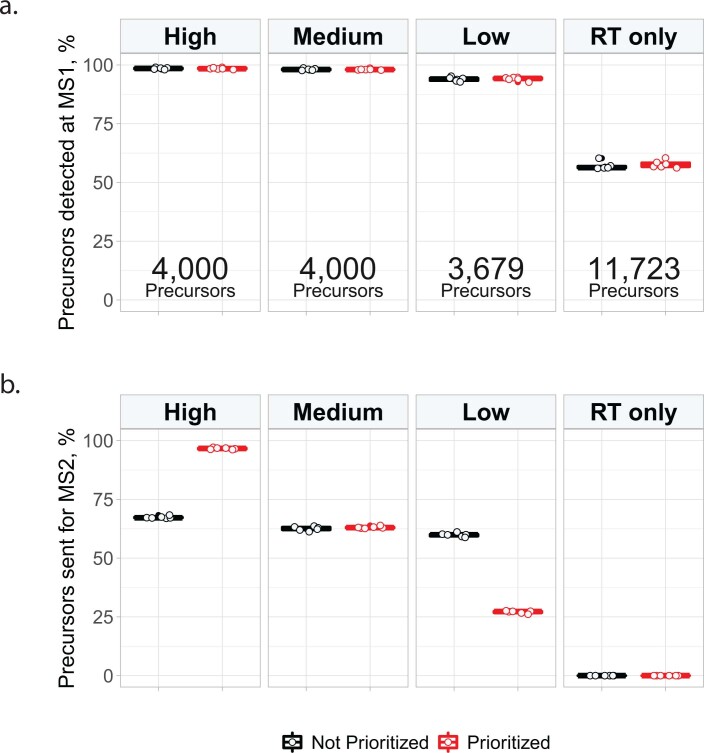
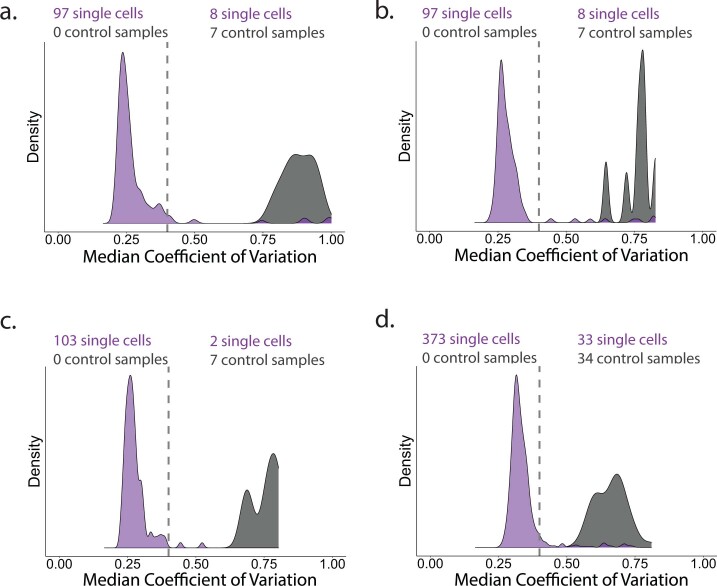
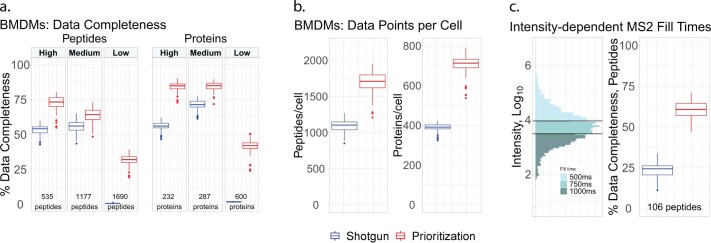
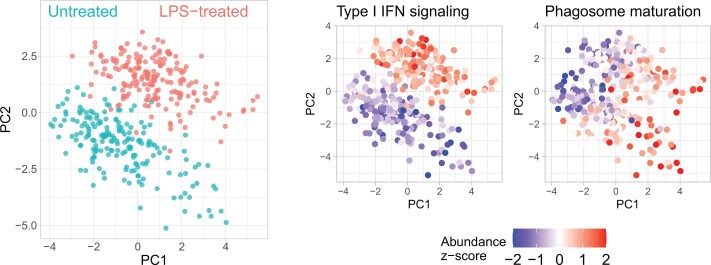
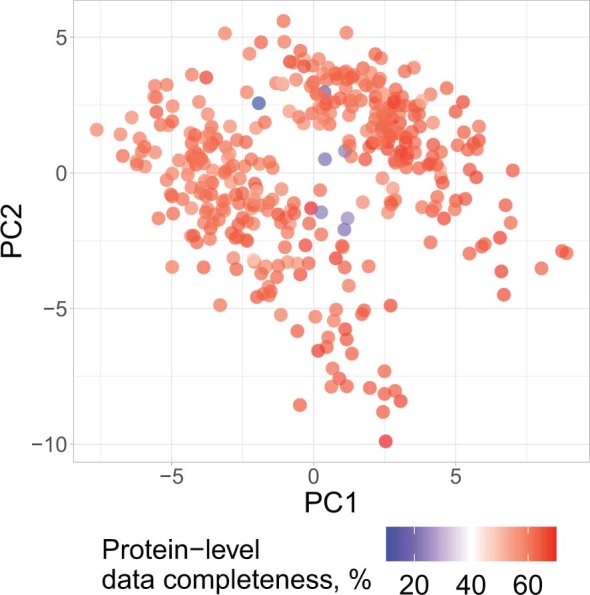
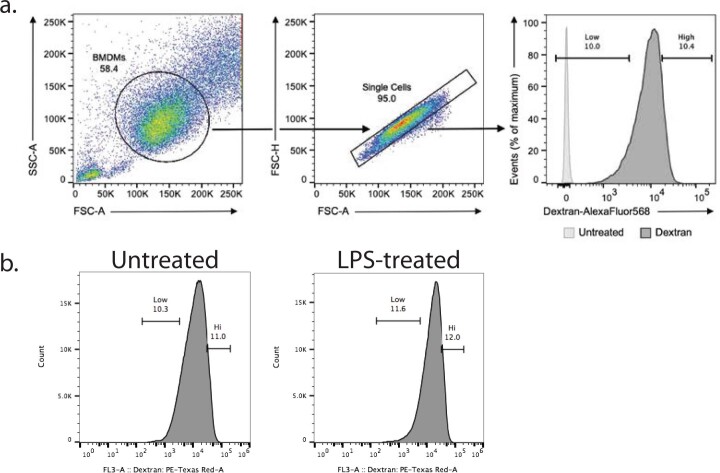
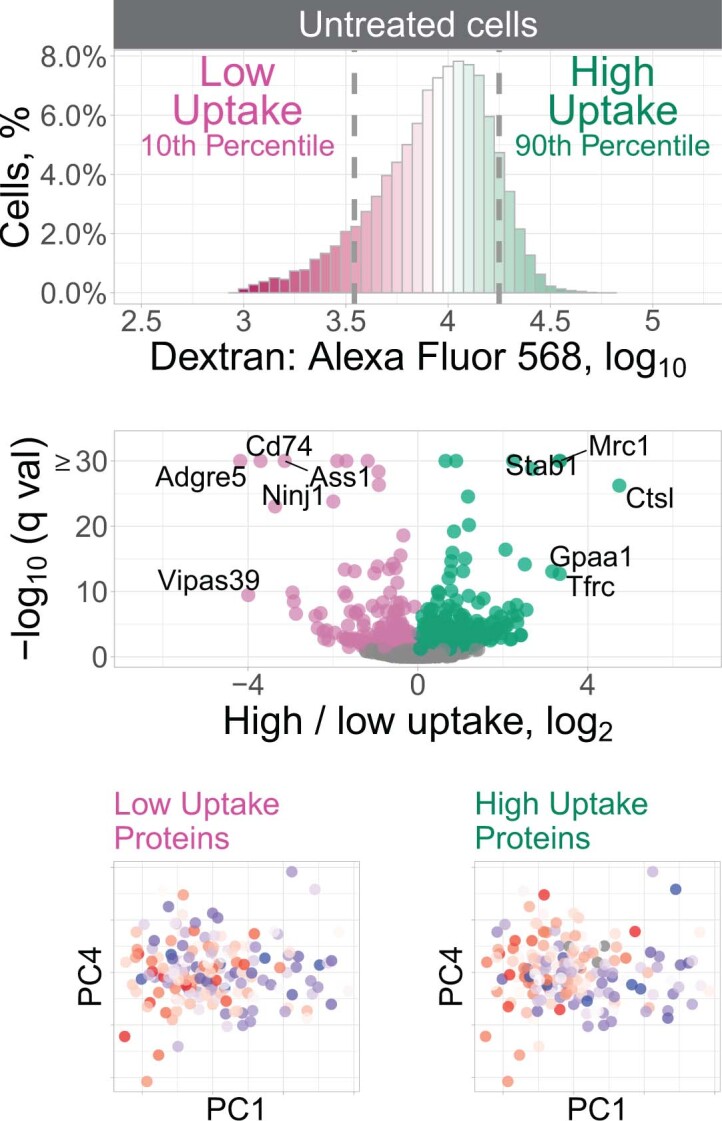
Major aims of single-cell proteomics include increasing the consistency, sensitivity and depth of protein quantification, especially for proteins and modifications of biological interest. Here, to simultaneously advance all these aims, we developed prioritized Single-Cell ProtEomics (pSCoPE). pSCoPE consistently analyzes thousands of prioritized peptides across all single cells (thus increasing data completeness) while maximizing instrument time spent analyzing identifiable peptides, thus increasing proteome depth. These strategies increased the sensitivity, data completeness and proteome coverage over twofold. The gains enabled quantifying protein variation in untreated and lipopolysaccharide-treated primary macrophages. Within each condition, proteins covaried within functional sets, including phagosome maturation and proton transport, similarly across both treatment conditions. This covariation is coupled to phenotypic variability in endocytic activity. pSCoPE also enabled quantifying proteolytic products, suggesting a gradient of cathepsin activities within a treatment condition. pSCoPE is freely available and widely applicable, especially for analyzing proteins of interest without sacrificing proteome coverage. Support for pSCoPE is available at http://scp.slavovlab.net/pSCoPE .
© 2023. The Author(s).
Conflict of interest statement
N.S. is a founding director and CEO of Parallel Squared Technology Institute, which is a nonprofit research institute.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
