

Targeting mitochondrial energetics reverses panobinostat- and marizomib-induced resistance in pediatric and adult high-grade gliomas
- PMID: 37014128
- PMCID: PMC10483615
- DOI: 10.1002/1878-0261.13427
Targeting mitochondrial energetics reverses panobinostat- and marizomib-induced resistance in pediatric and adult high-grade gliomas
Abstract
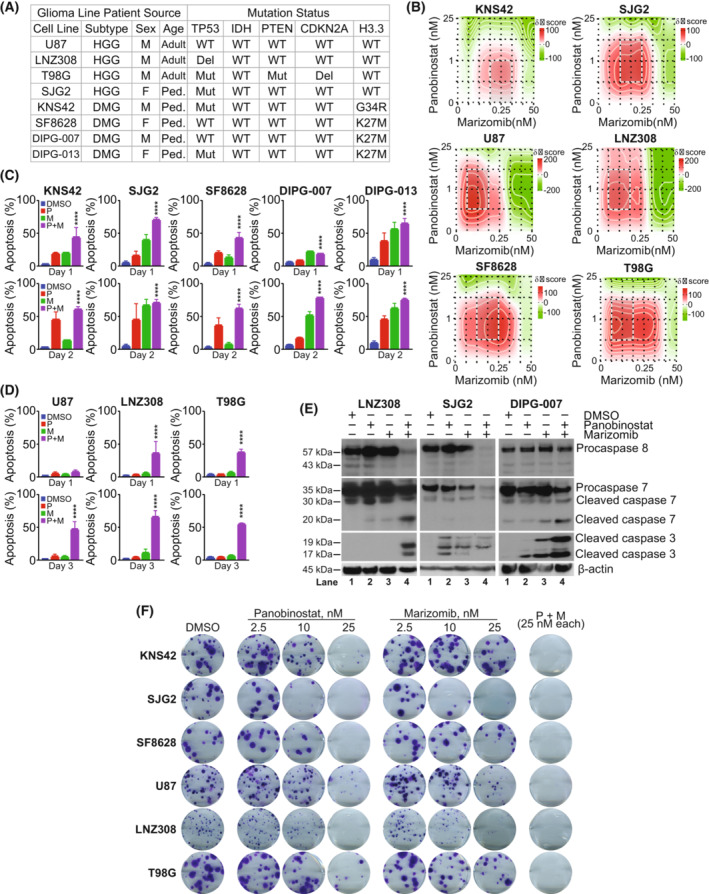
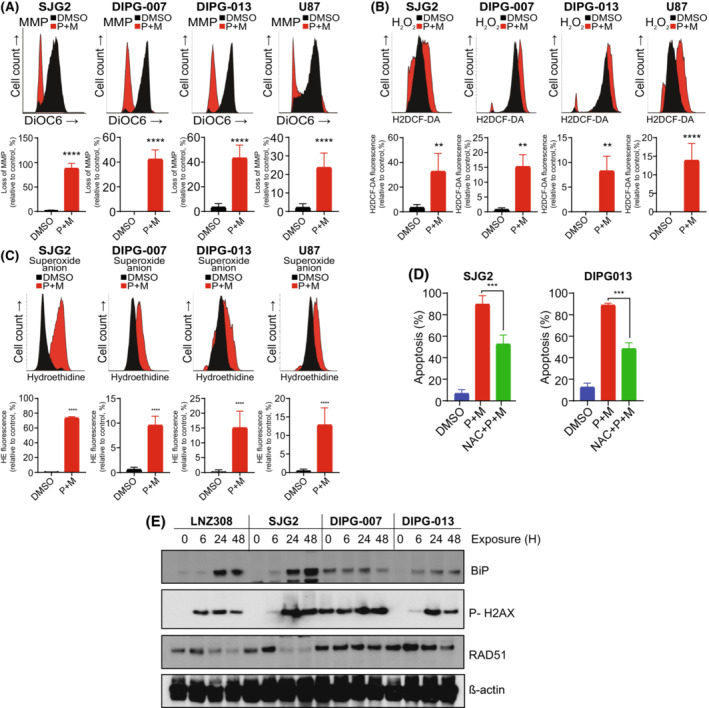
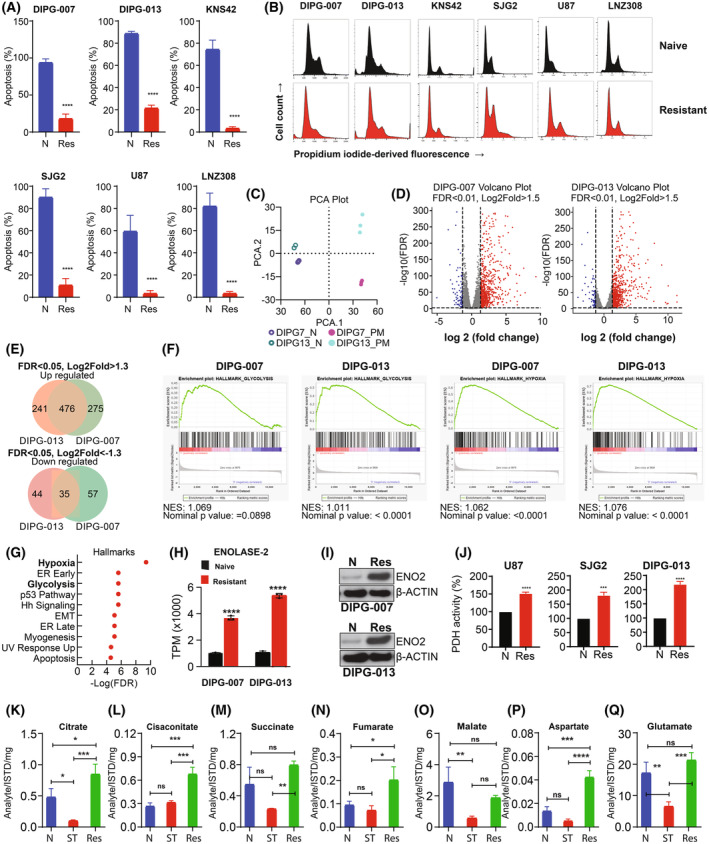
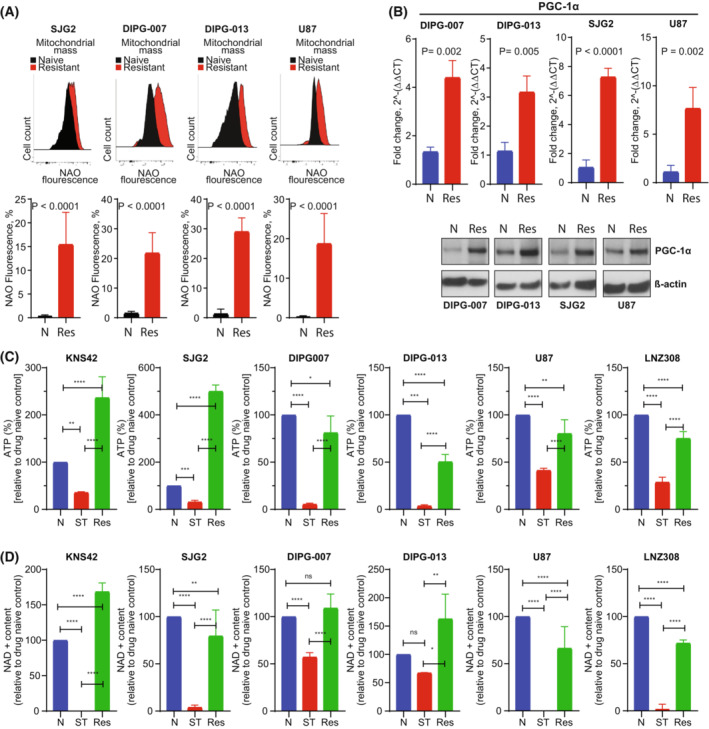
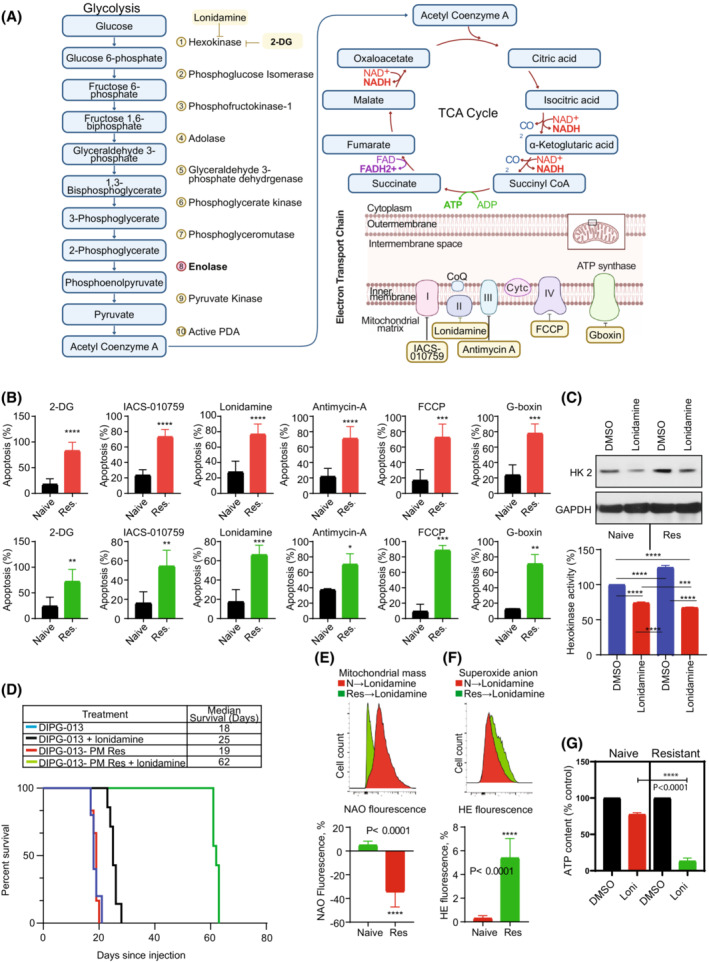
In previous studies, we demonstrated that panobinostat, a histone deacetylase inhibitor, and bortezomib, a proteasomal inhibitor, displayed synergistic therapeutic activity against pediatric and adult high-grade gliomas. Despite the remarkable initial response to this combination, resistance emerged. Here, in this study, we aimed to investigate the molecular mechanisms underlying the anticancer effects of panobinostat and marizomib, a brain-penetrant proteasomal inhibitor, and the potential for exploitable vulnerabilities associated with acquired resistance. RNA sequencing followed by gene set enrichment analysis (GSEA) was employed to compare the molecular signatures enriched in resistant compared with drug-naïve cells. The levels of adenosine 5'-triphosphate (ATP), nicotinamide adenine dinucleotide (NAD)+ content, hexokinase activity, and tricarboxylic acid (TCA) cycle metabolites required for oxidative phosphorylation to meet their bioenergetic needs were analyzed. Here, we report that panobinostat and marizomib significantly depleted ATP and NAD+ content, increased mitochondrial permeability and reactive oxygen species generation, and promoted apoptosis in pediatric and adult glioma cell lines at initial treatment. However, resistant cells exhibited increased levels of TCA cycle metabolites, which required for oxidative phosphorylation to meet their bioenergetic needs. Therefore, we targeted glycolysis and the electron transport chain (ETC) with small molecule inhibitors, which displayed substantial efficacy, suggesting that resistant cell survival is dependent on glycolytic and ETC complexes. To verify these observations in vivo, lonidamine, an inhibitor of glycolysis and mitochondrial function, was chosen. We produced two diffuse intrinsic pontine glioma (DIPG) models, and lonidamine treatment significantly increased median survival in both models, with particularly dramatic effects in panobinostat- and marizomib-resistant cells. These data provide new insights into mechanisms of treatment resistance in gliomas.
Keywords: OXPHOS; glioma; marizomib; mitochondria; panobinostat; resistance.
© 2023 The Authors. Molecular Oncology published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Wick W, Kessler T. New glioblastoma heterogeneity atlas – a shared resource. Nat Rev Neurol. 2018;14:453–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
