

Identification of RNA Virus-Derived RdRp Sequences in Publicly Available Transcriptomic Data Sets
- PMID: 37014783
- PMCID: PMC10101049
- DOI: 10.1093/molbev/msad060
Identification of RNA Virus-Derived RdRp Sequences in Publicly Available Transcriptomic Data Sets
Abstract
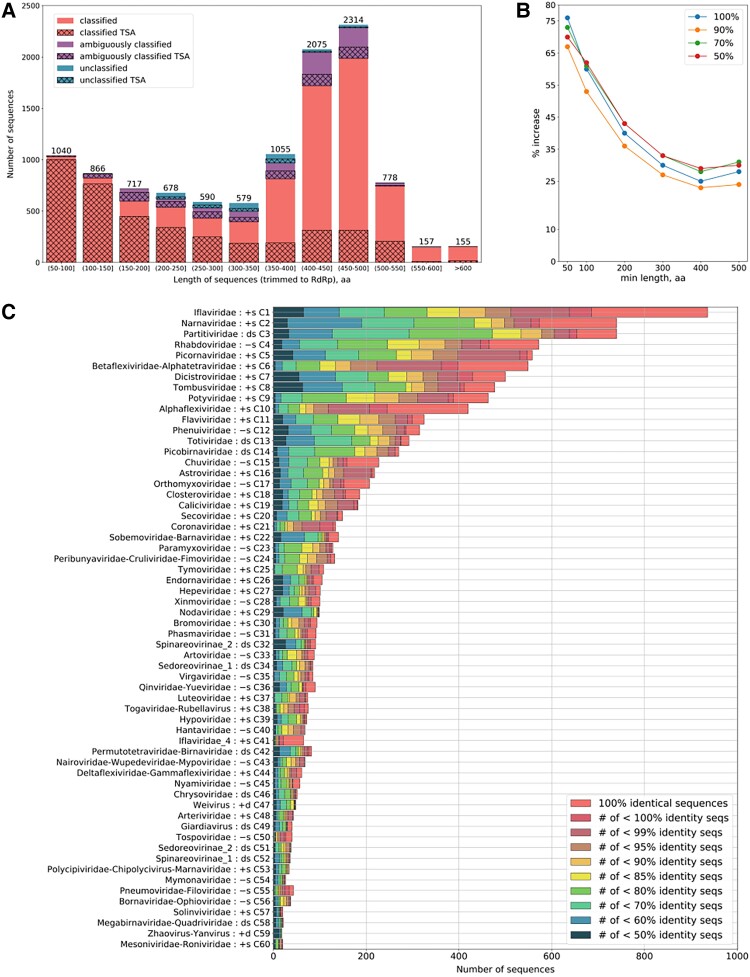
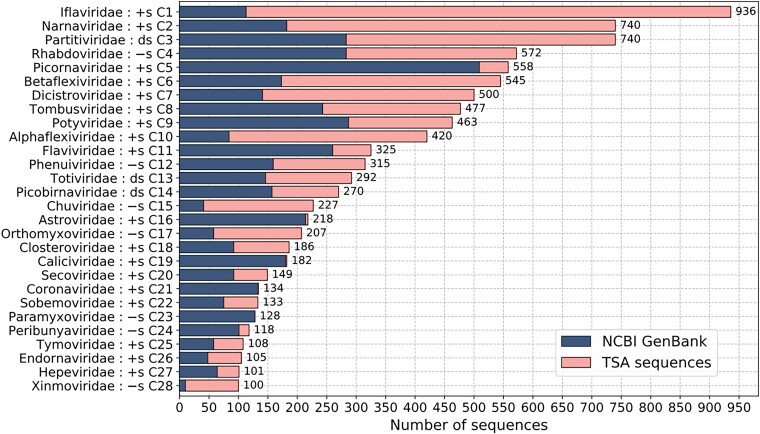
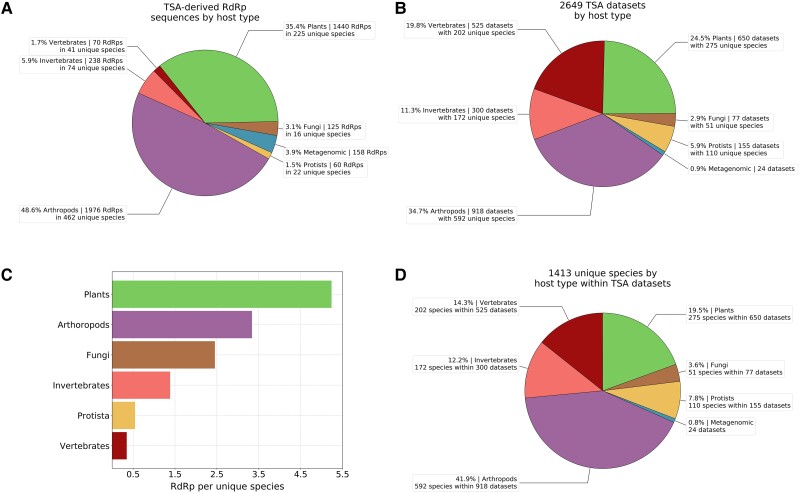
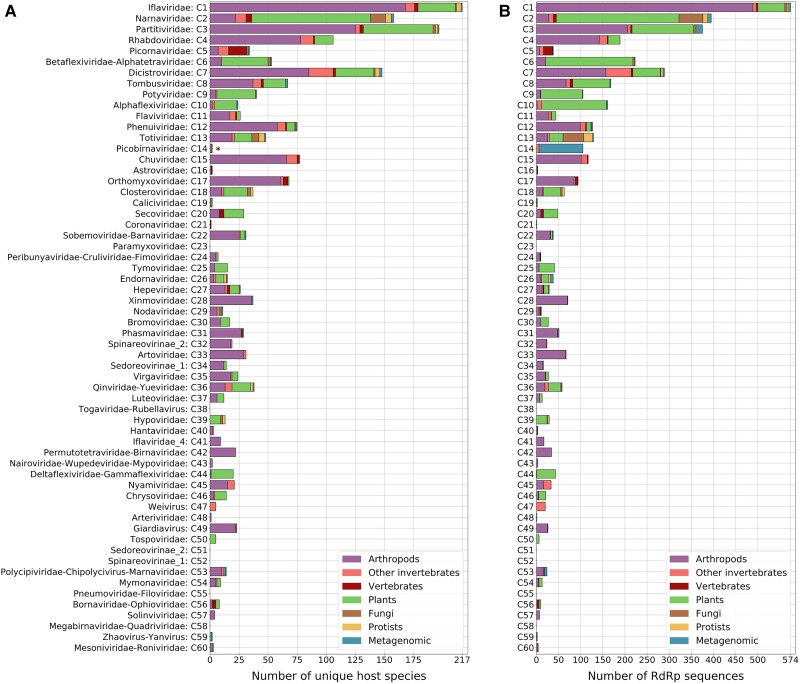
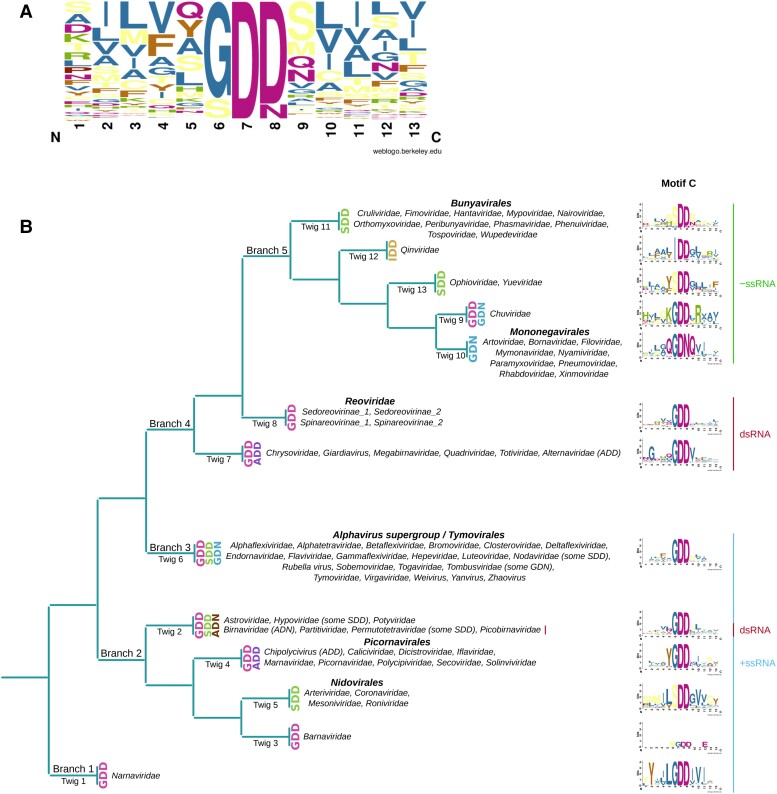
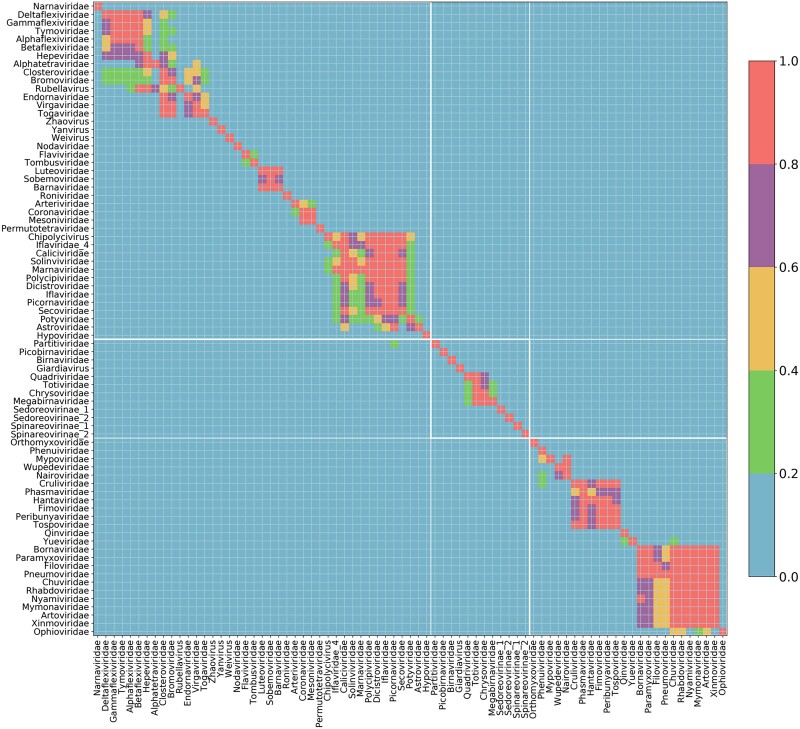
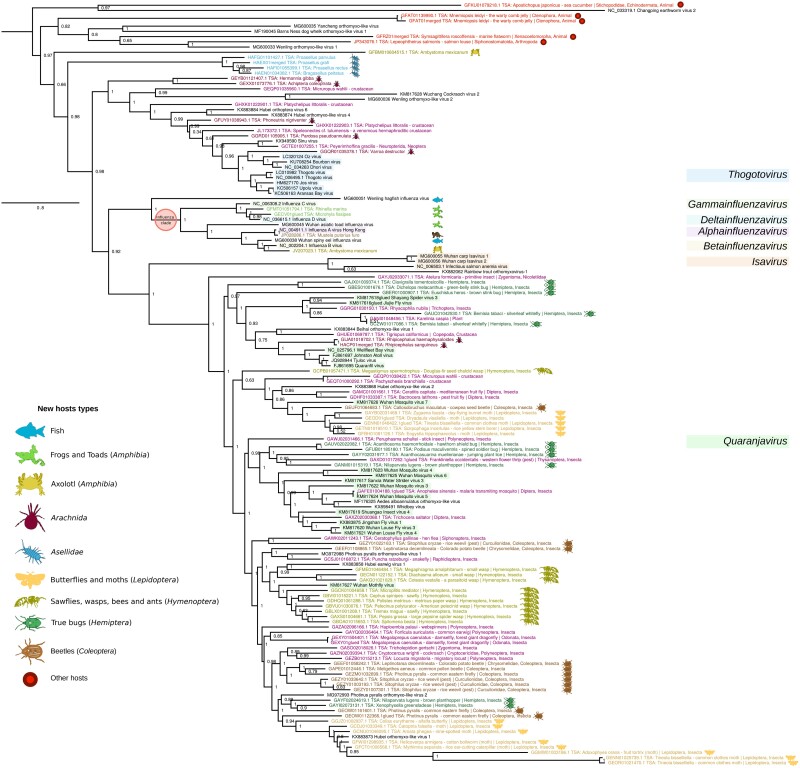
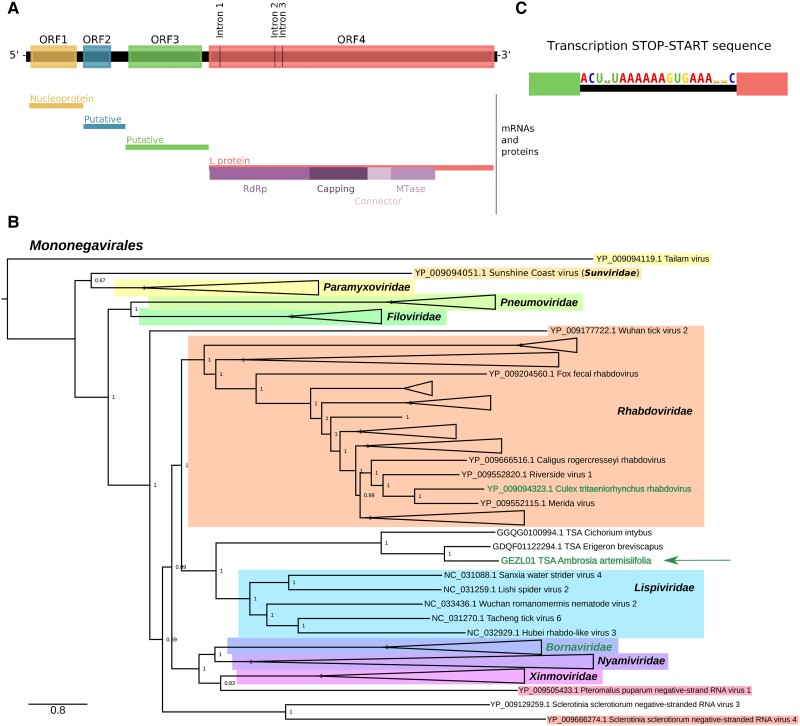
RNA viruses are abundant and highly diverse and infect all or most eukaryotic organisms. However, only a tiny fraction of the number and diversity of RNA virus species have been catalogued. To cost-effectively expand the diversity of known RNA virus sequences, we mined publicly available transcriptomic data sets. We developed 77 family-level Hidden Markov Model profiles for the viral RNA-dependent RNA polymerase (RdRp)-the only universal "hallmark" gene of RNA viruses. By using these to search the National Center for Biotechnology Information Transcriptome Shotgun Assembly database, we identified 5,867 contigs encoding RNA virus RdRps or fragments thereof and analyzed their diversity, taxonomic classification, phylogeny, and host associations. Our study expands the known diversity of RNA viruses, and the 77 curated RdRp Profile Hidden Markov Models provide a useful resource for the virus discovery community.
Keywords: Orthomyxoviridae; RNA virus; RdRp; pHMM; splicing; virus discovery.
© The Author(s) 2023. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
References
-
- Aiewsakun P, Katzourakis A. 2015. Endogenous viruses: connecting recent and ancient viral evolution. Virology 479–480:26–37. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. - PubMed
-
- Arjona-Lopez JM, Telengech P, Jamal A, Hisano S, Kondo H, Yelin MD, Arjona-Girona I, Kanematsu S, Lopez-Herrera CJ, Suzuki N. 2018. Novel, diverse RNA viruses from Mediterranean isolates of the phytopathogenic fungus, Rosellinia necatrix: insights into evolutionary biology of fungal viruses. Environ Microbiol. 20(4):1464–1483. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
