

Developing an appropriate evolutionary baseline model for the study of SARS-CoV-2 patient samples
- PMID: 37018331
- PMCID: PMC10075409
- DOI: 10.1371/journal.ppat.1011265
Developing an appropriate evolutionary baseline model for the study of SARS-CoV-2 patient samples
Abstract
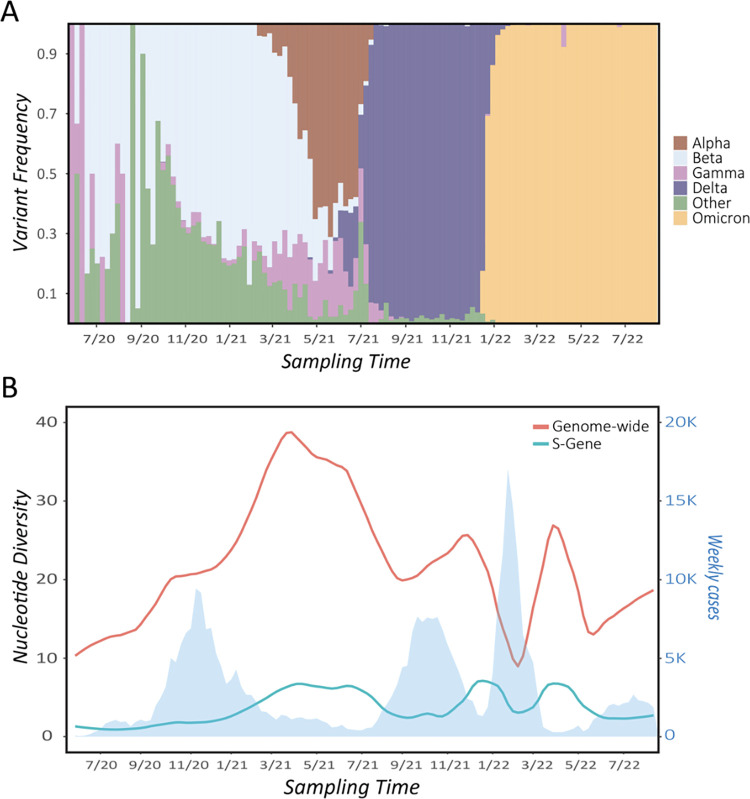
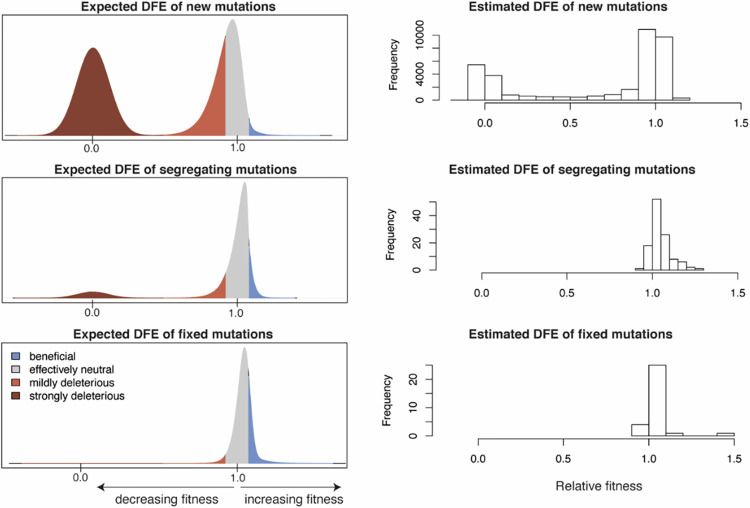
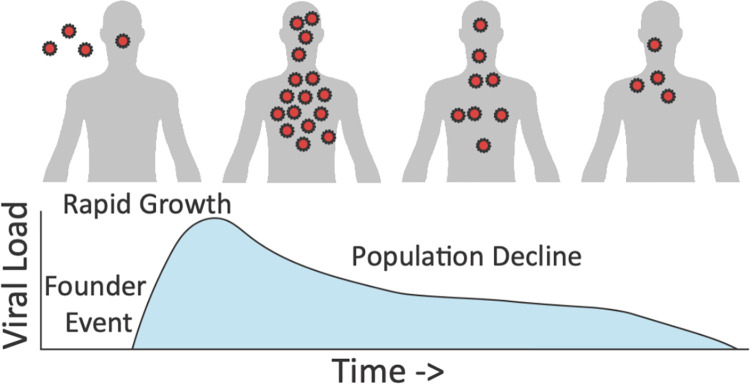
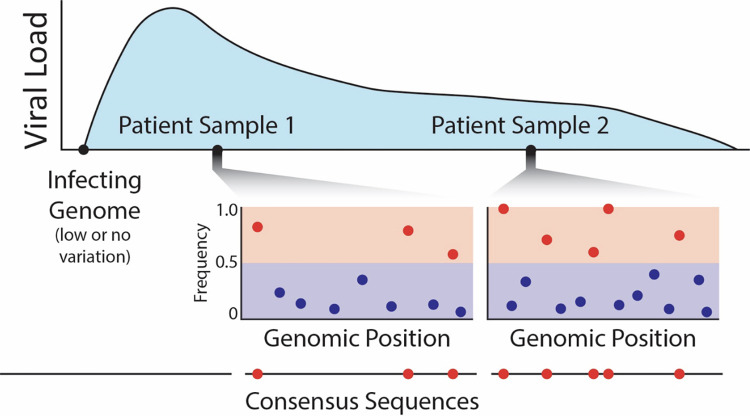
Over the past 3 years, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has spread through human populations in several waves, resulting in a global health crisis. In response, genomic surveillance efforts have proliferated in the hopes of tracking and anticipating the evolution of this virus, resulting in millions of patient isolates now being available in public databases. Yet, while there is a tremendous focus on identifying newly emerging adaptive viral variants, this quantification is far from trivial. Specifically, multiple co-occurring and interacting evolutionary processes are constantly in operation and must be jointly considered and modeled in order to perform accurate inference. We here outline critical individual components of such an evolutionary baseline model-mutation rates, recombination rates, the distribution of fitness effects, infection dynamics, and compartmentalization-and describe the current state of knowledge pertaining to the related parameters of each in SARS-CoV-2. We close with a series of recommendations for future clinical sampling, model construction, and statistical analysis.
Copyright: © 2023 Terbot et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
