

Etiology of oncogenic fusions in 5,190 childhood cancers and its clinical and therapeutic implication
- PMID: 37019972
- PMCID: PMC10076316
- DOI: 10.1038/s41467-023-37438-4
Etiology of oncogenic fusions in 5,190 childhood cancers and its clinical and therapeutic implication
Abstract
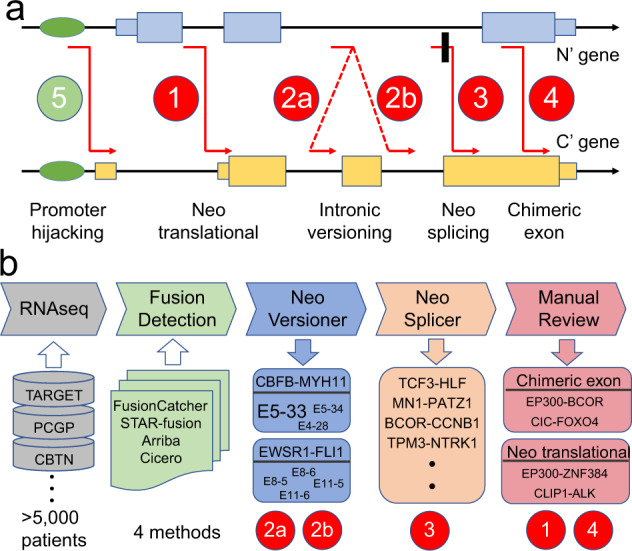
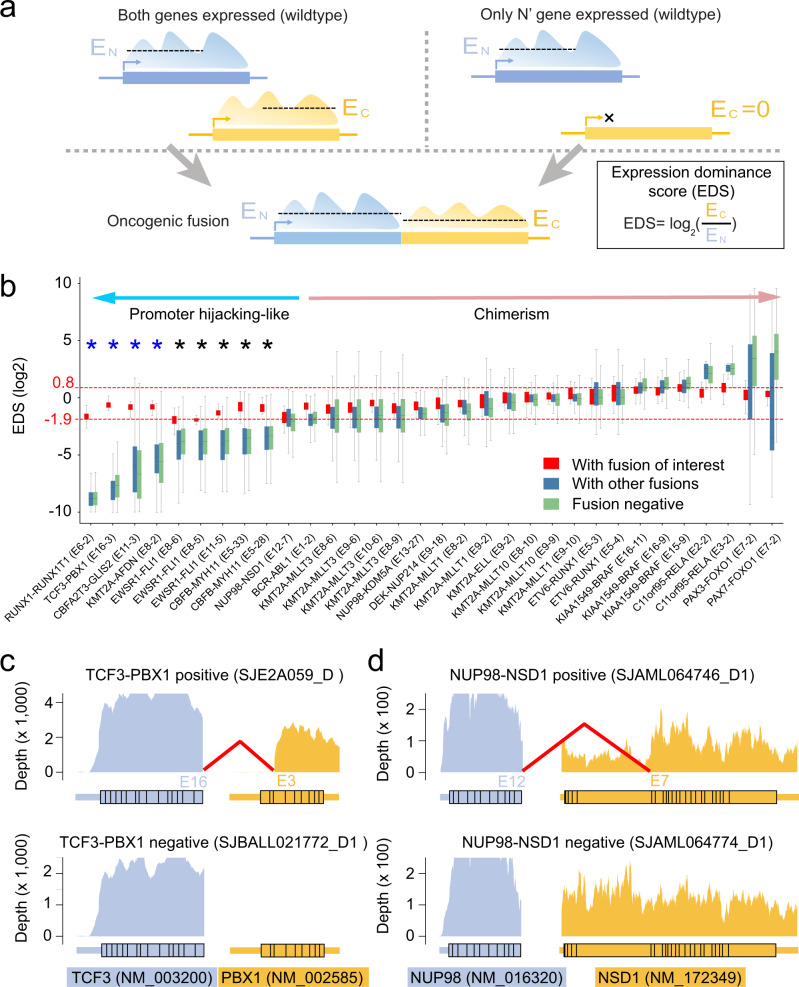
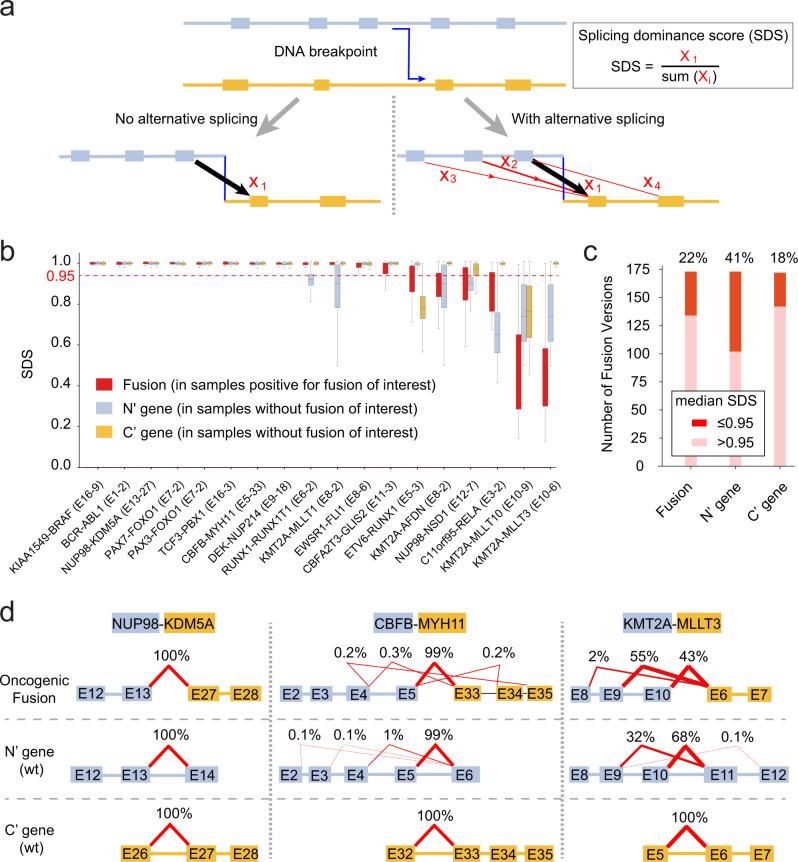
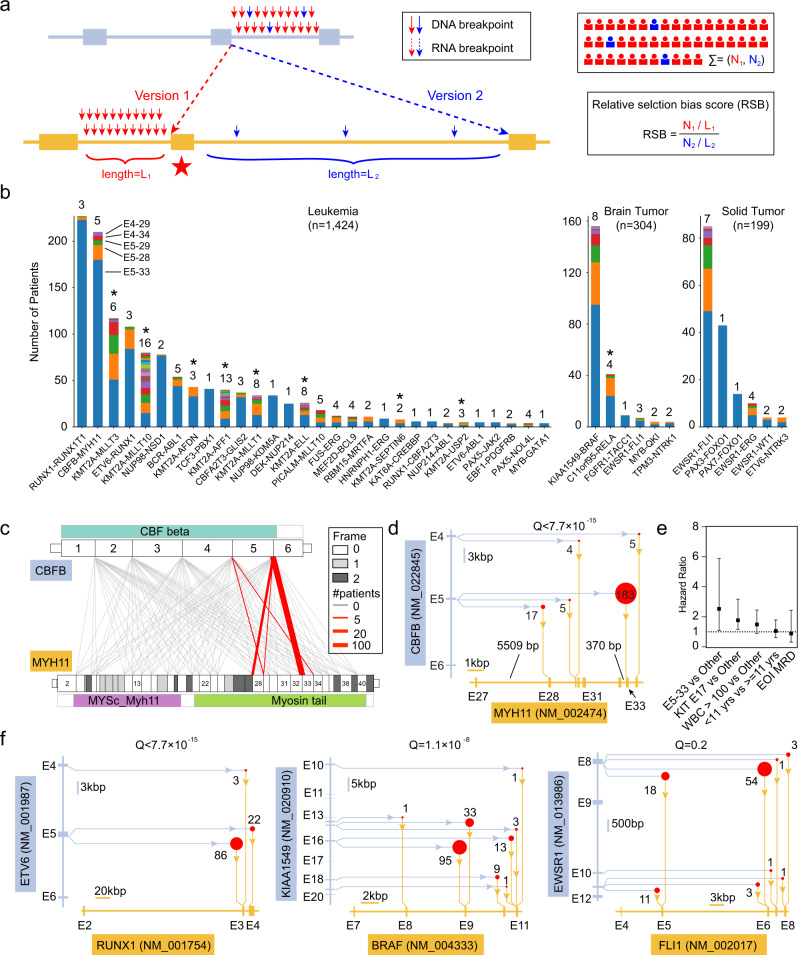
Oncogenic fusions formed through chromosomal rearrangements are hallmarks of childhood cancer that define cancer subtype, predict outcome, persist through treatment, and can be ideal therapeutic targets. However, mechanistic understanding of the etiology of oncogenic fusions remains elusive. Here we report a comprehensive detection of 272 oncogenic fusion gene pairs by using tumor transcriptome sequencing data from 5190 childhood cancer patients. We identify diverse factors, including translation frame, protein domain, splicing, and gene length, that shape the formation of oncogenic fusions. Our mathematical modeling reveals a strong link between differential selection pressure and clinical outcome in CBFB-MYH11. We discover 4 oncogenic fusions, including RUNX1-RUNX1T1, TCF3-PBX1, CBFA2T3-GLIS2, and KMT2A-AFDN, with promoter-hijacking-like features that may offer alternative strategies for therapeutic targeting. We uncover extensive alternative splicing in oncogenic fusions including KMT2A-MLLT3, KMT2A-MLLT10, C11orf95-RELA, NUP98-NSD1, KMT2A-AFDN and ETV6-RUNX1. We discover neo splice sites in 18 oncogenic fusion gene pairs and demonstrate that such splice sites confer therapeutic vulnerability for etiology-based genome editing. Our study reveals general principles on the etiology of oncogenic fusions in childhood cancer and suggests profound clinical implications including etiology-based risk stratification and genome-editing-based therapeutics.
© 2023. The Author(s).
Conflict of interest statement
X.M. is a named inventor on a pending patent application based in part on the research disclosed in this manuscript. The authors declare that they have no competing interests.
Figures

References
-
- Nowell, P. & Hungerford, D. A minute chromosome in human chronic granulocytic leukemia [abstract]. Science132, 1497 (1960).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
