

De novo design of modular peptide-binding proteins by superhelical matching
- PMID: 37020023
- PMCID: PMC10115654
- DOI: 10.1038/s41586-023-05909-9
De novo design of modular peptide-binding proteins by superhelical matching
Abstract
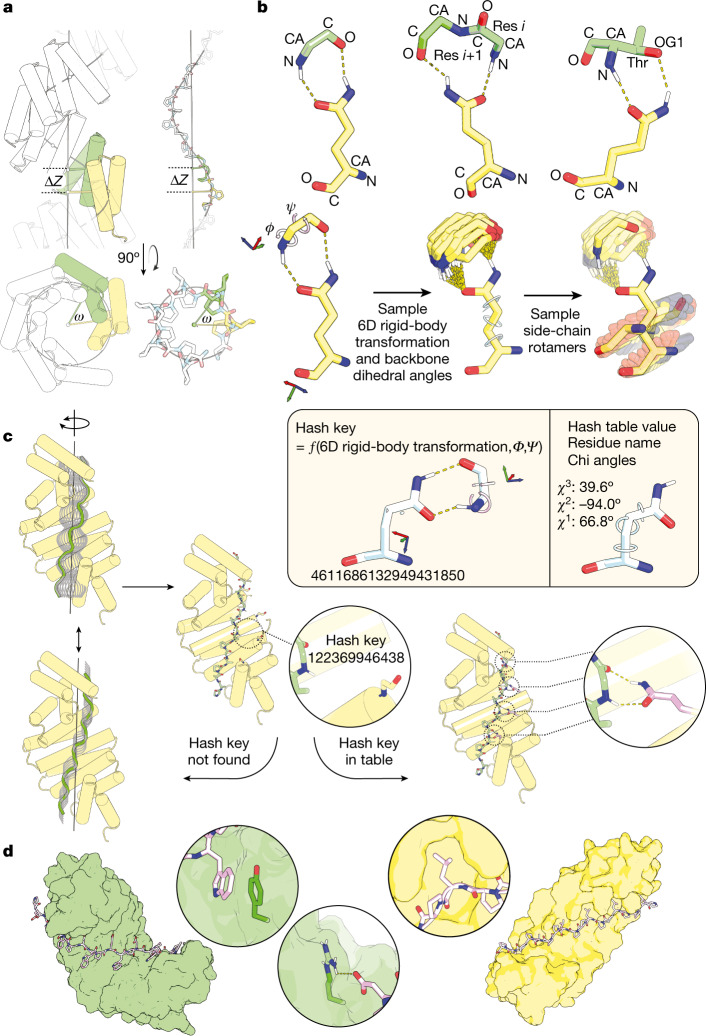
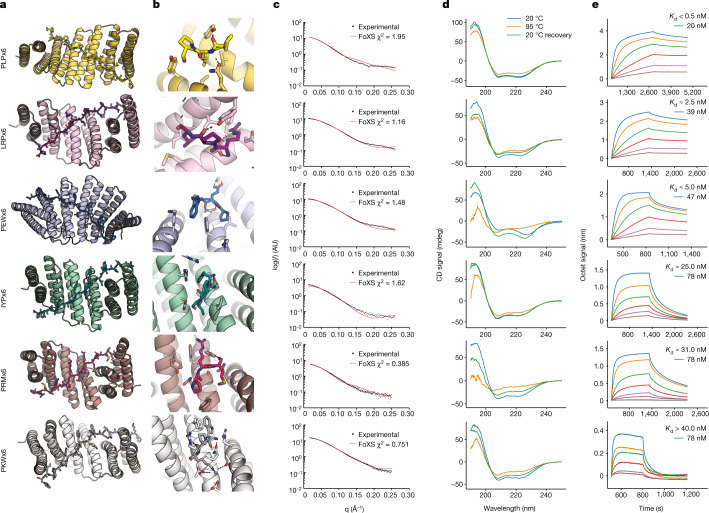
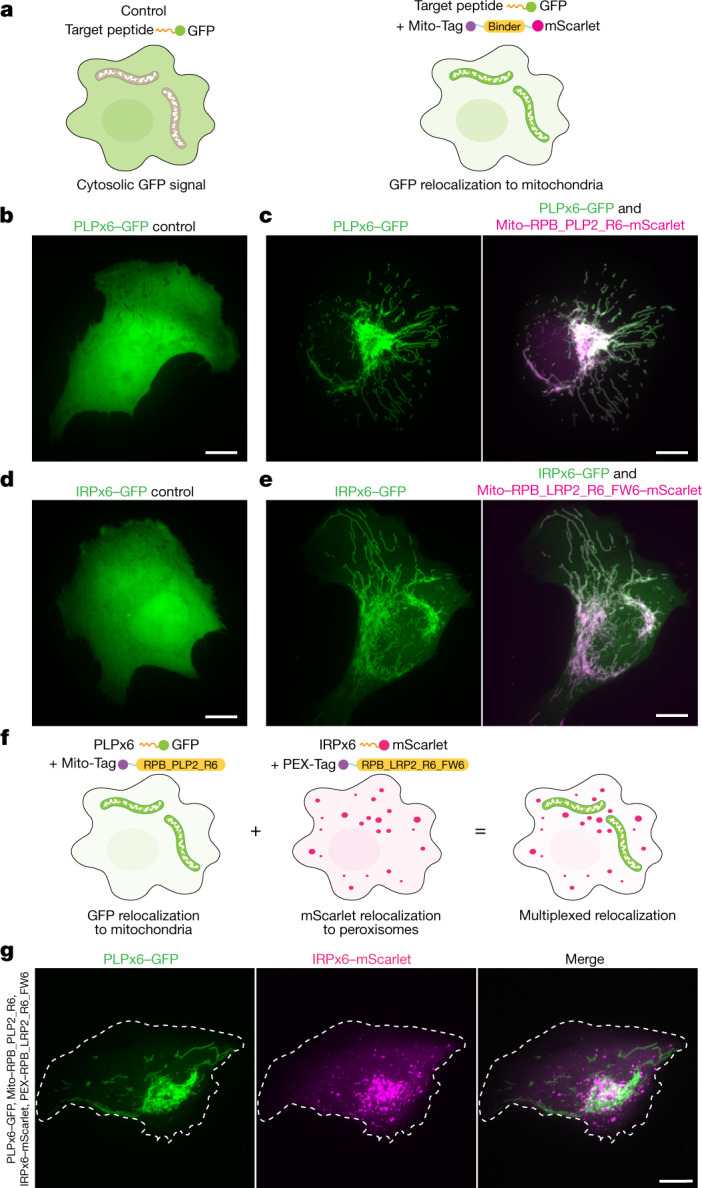
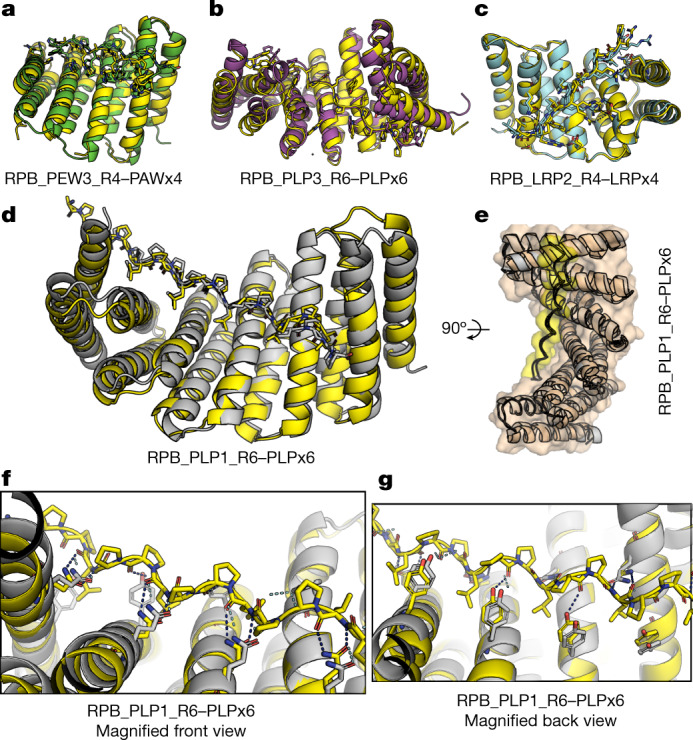
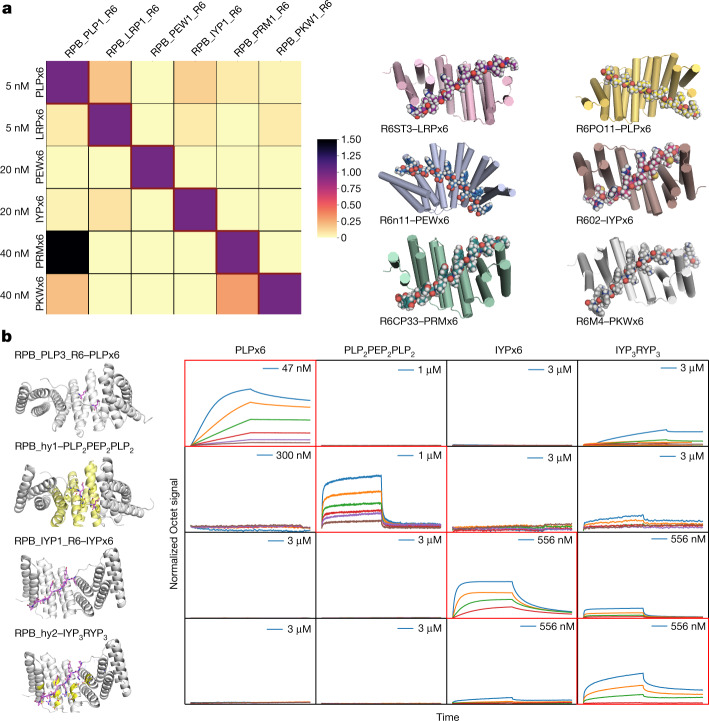
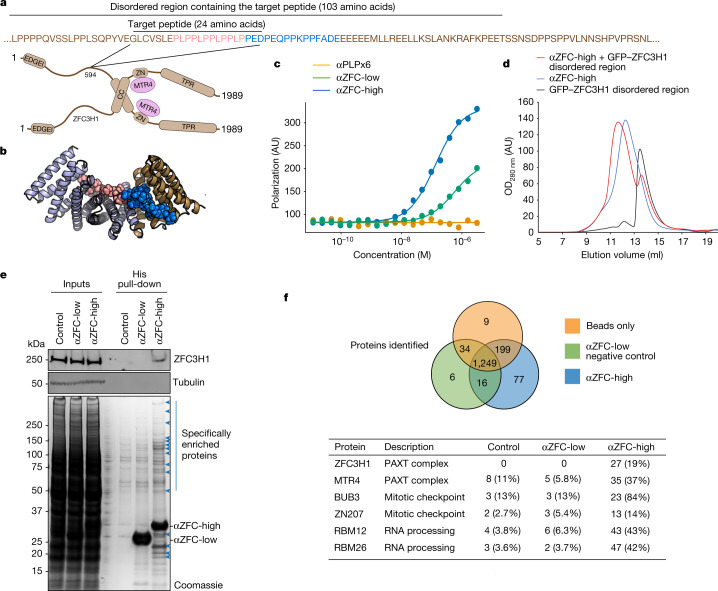
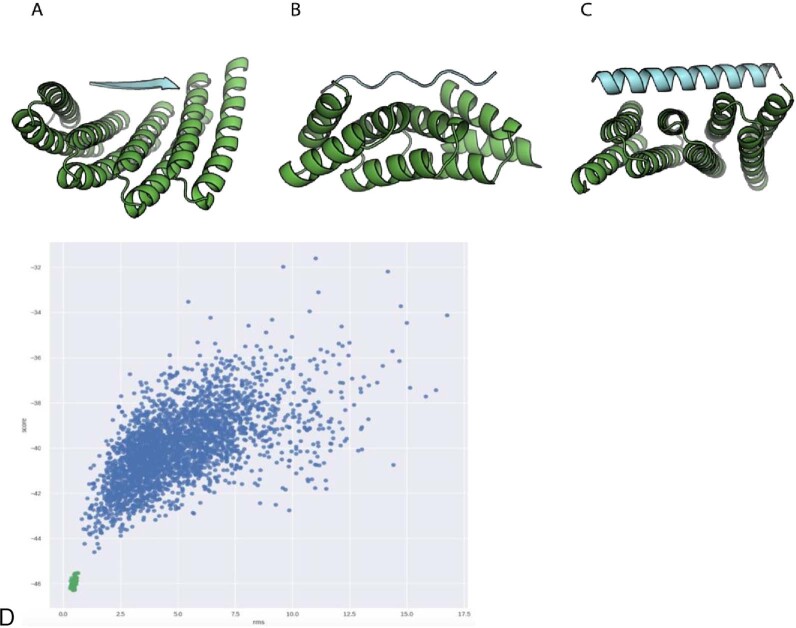
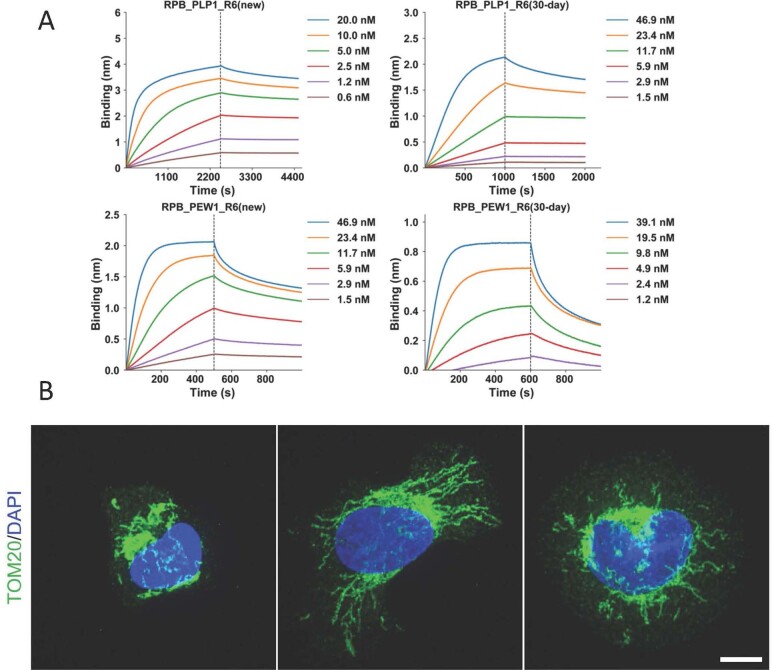
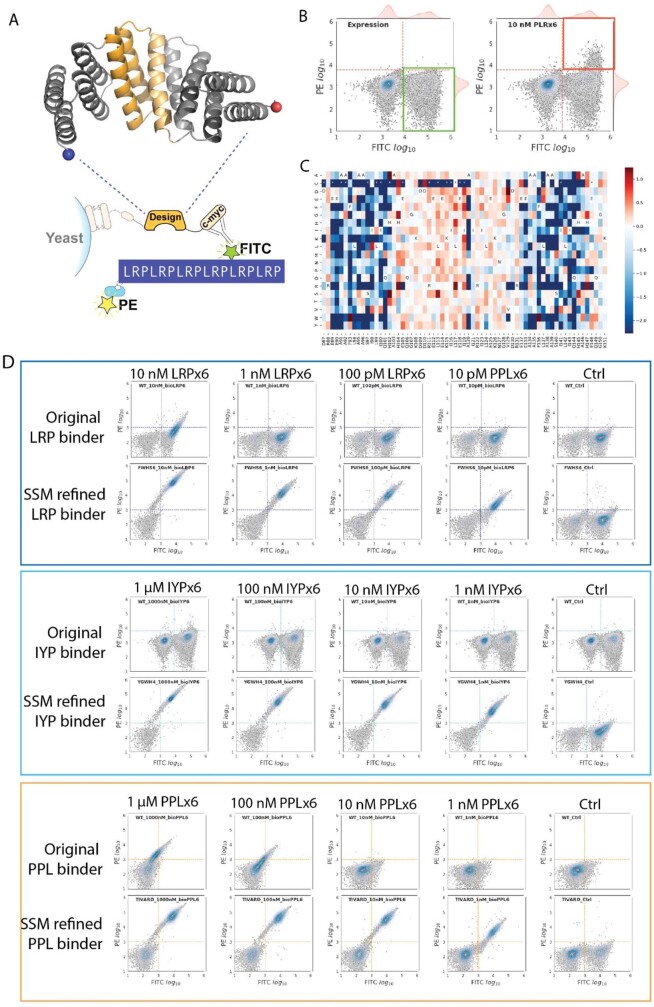
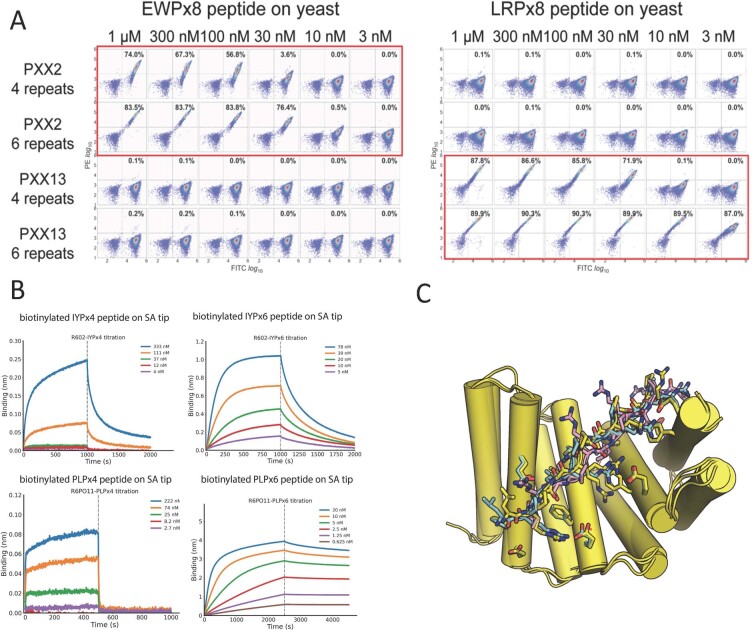
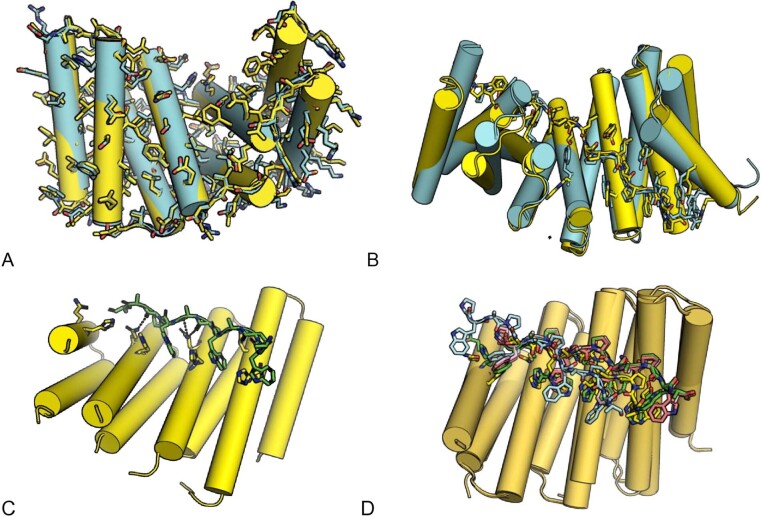
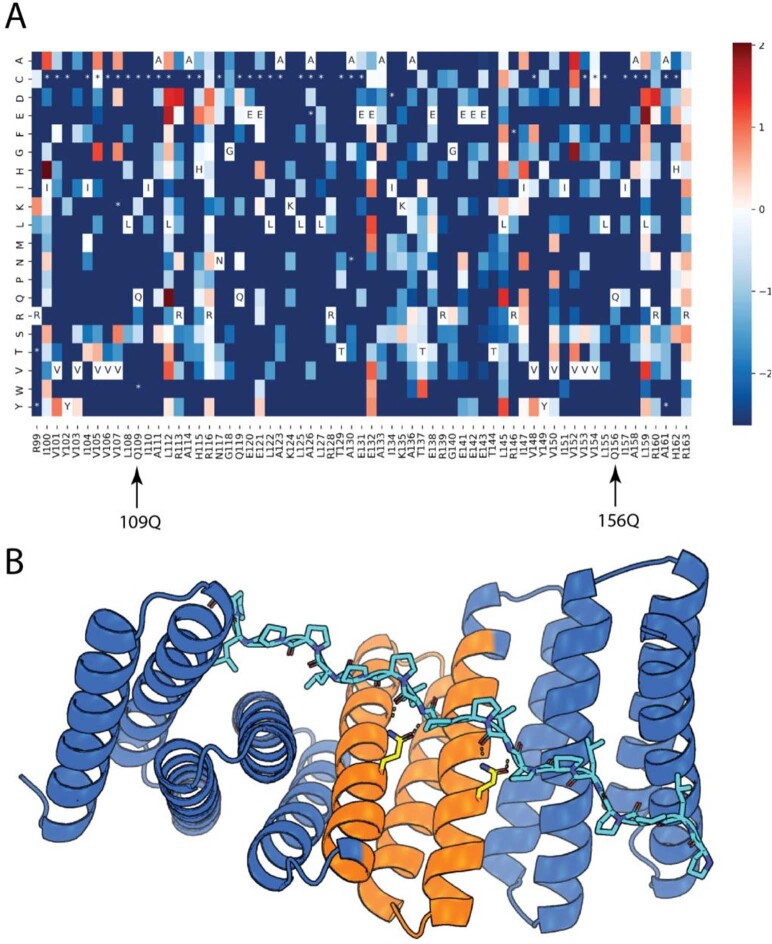
General approaches for designing sequence-specific peptide-binding proteins would have wide utility in proteomics and synthetic biology. However, designing peptide-binding proteins is challenging, as most peptides do not have defined structures in isolation, and hydrogen bonds must be made to the buried polar groups in the peptide backbone1-3. Here, inspired by natural and re-engineered protein-peptide systems4-11, we set out to design proteins made out of repeating units that bind peptides with repeating sequences, with a one-to-one correspondence between the repeat units of the protein and those of the peptide. We use geometric hashing to identify protein backbones and peptide-docking arrangements that are compatible with bidentate hydrogen bonds between the side chains of the protein and the peptide backbone12. The remainder of the protein sequence is then optimized for folding and peptide binding. We design repeat proteins to bind to six different tripeptide-repeat sequences in polyproline II conformations. The proteins are hyperstable and bind to four to six tandem repeats of their tripeptide targets with nanomolar to picomolar affinities in vitro and in living cells. Crystal structures reveal repeating interactions between protein and peptide interactions as designed, including ladders of hydrogen bonds from protein side chains to peptide backbones. By redesigning the binding interfaces of individual repeat units, specificity can be achieved for non-repeating peptide sequences and for disordered regions of native proteins.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests, except as follows. K.W., H.B., D.R.H., T.J.B., K.E.M., T.J.S., T.E.M., Y.-T.C., R.R., G.B., D.C.E., L.S., E.D., D.A.S., W.S., I.G. and D.B. are co-inventors on a patent application entitled ‘De novo designed modular peptide binding proteins by superhelical matching’ (63/381,109, filed 26 October 2022).
Figures
References
-
- Andrade, M. A., Petosa, C., O’Donoghue, S. I., Müller, C. W. & Bork, P. Comparison of ARM and HEAT protein repeats. J. Mol. Biol.309, 1–18 (2001). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
