

The role of immunometabolism in macrophage polarization and its impact on acute lung injury/acute respiratory distress syndrome
- PMID: 37020557
- PMCID: PMC10067752
- DOI: 10.3389/fimmu.2023.1117548
The role of immunometabolism in macrophage polarization and its impact on acute lung injury/acute respiratory distress syndrome
Abstract
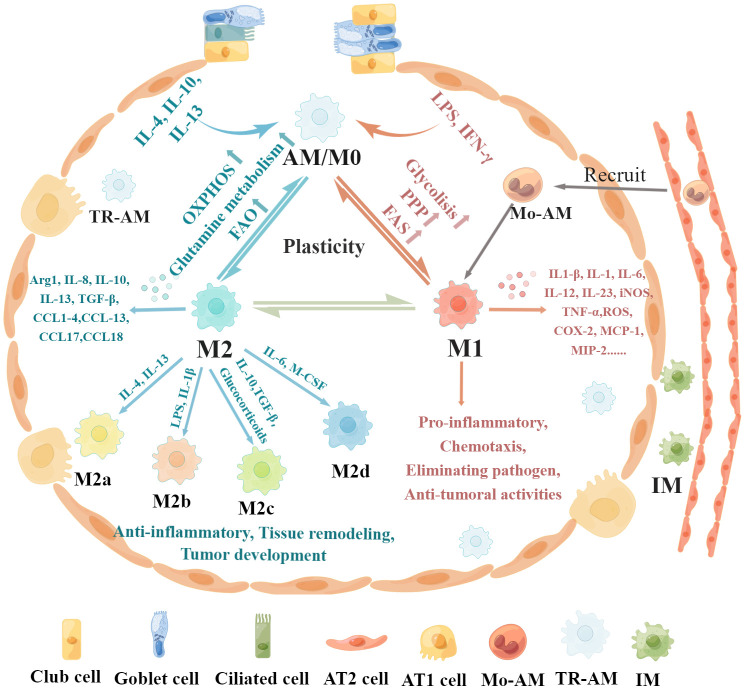
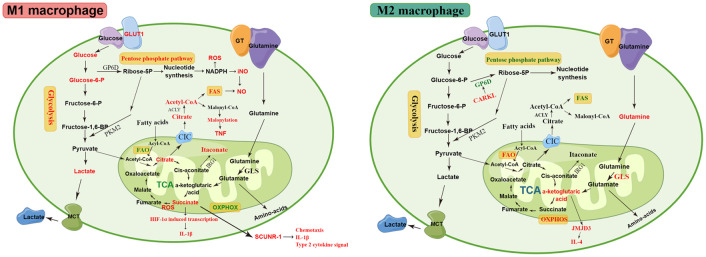
Lung macrophages constitute the first line of defense against airborne particles and microbes and are key to maintaining pulmonary immune homeostasis. There is increasing evidence suggesting that macrophages also participate in the pathogenesis of acute lung injury (ALI)/acute respiratory distress syndrome (ARDS), including the modulation of inflammatory responses and the repair of damaged lung tissues. The diversity of their functions may be attributed to their polarized states. Classically activated or inflammatory (M1) macrophages and alternatively activated or anti-inflammatory (M2) macrophages are the two main polarized macrophage phenotypes. The precise regulatory mechanism of macrophage polarization is a complex process that is not completely understood. A growing body of literature on immunometabolism has demonstrated the essential role of immunometabolism and its metabolic intermediates in macrophage polarization. In this review, we summarize macrophage polarization phenotypes, the role of immunometabolism, and its metabolic intermediates in macrophage polarization and ALI/ARDS, which may represent a new target and therapeutic direction.
Keywords: acute lung injury; acute respiratory distress syndrome; immunometabolism; macrophage polarization; metabolic reprogramming; polarization regulation.
Copyright © 2023 Wang, Wang, Zhang, Ma, Tong and Fan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Comment in
-
Editorial: Cellular and systemic interplay of metabolism and inflammation in the pathogenesis of lung diseases.Front Immunol. 2024 Jan 17;14:1352304. doi: 10.3389/fimmu.2023.1352304. eCollection 2023. Front Immunol. 2024. PMID: 38299152 Free PMC article. No abstract available.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources