

Mitochondria in health, disease, and aging
- PMID: 37021870
- PMCID: PMC10393386
- DOI: 10.1152/physrev.00058.2021
Mitochondria in health, disease, and aging
Abstract
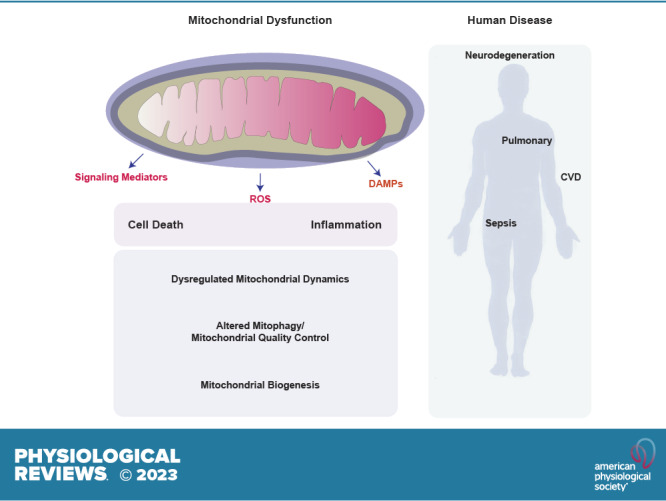
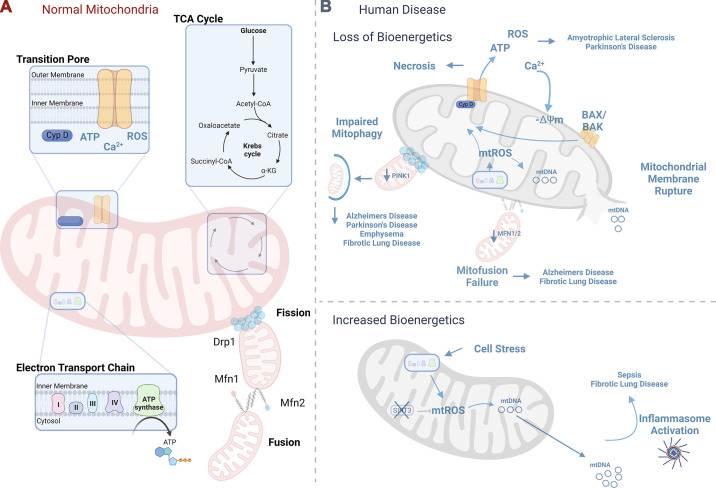
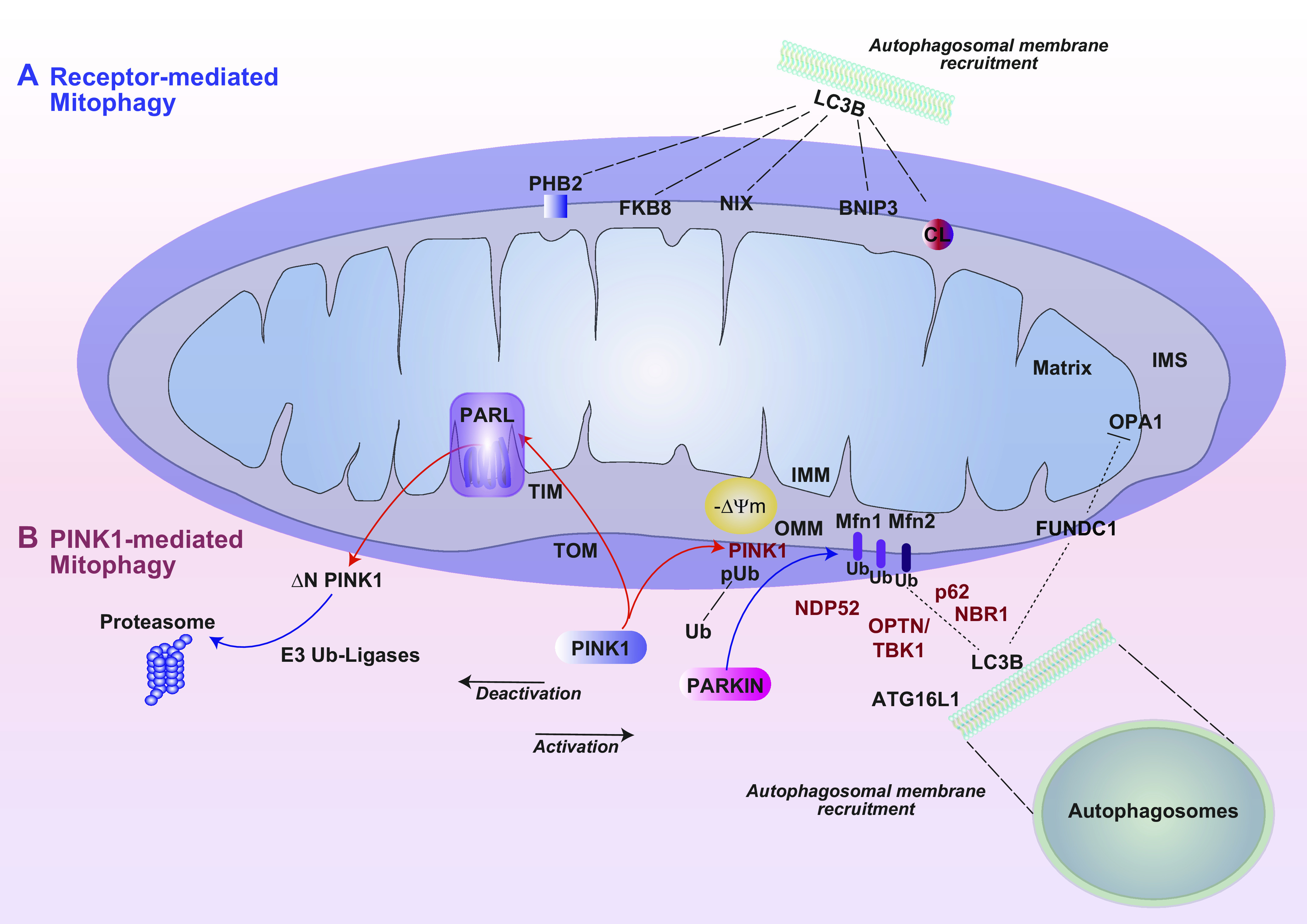
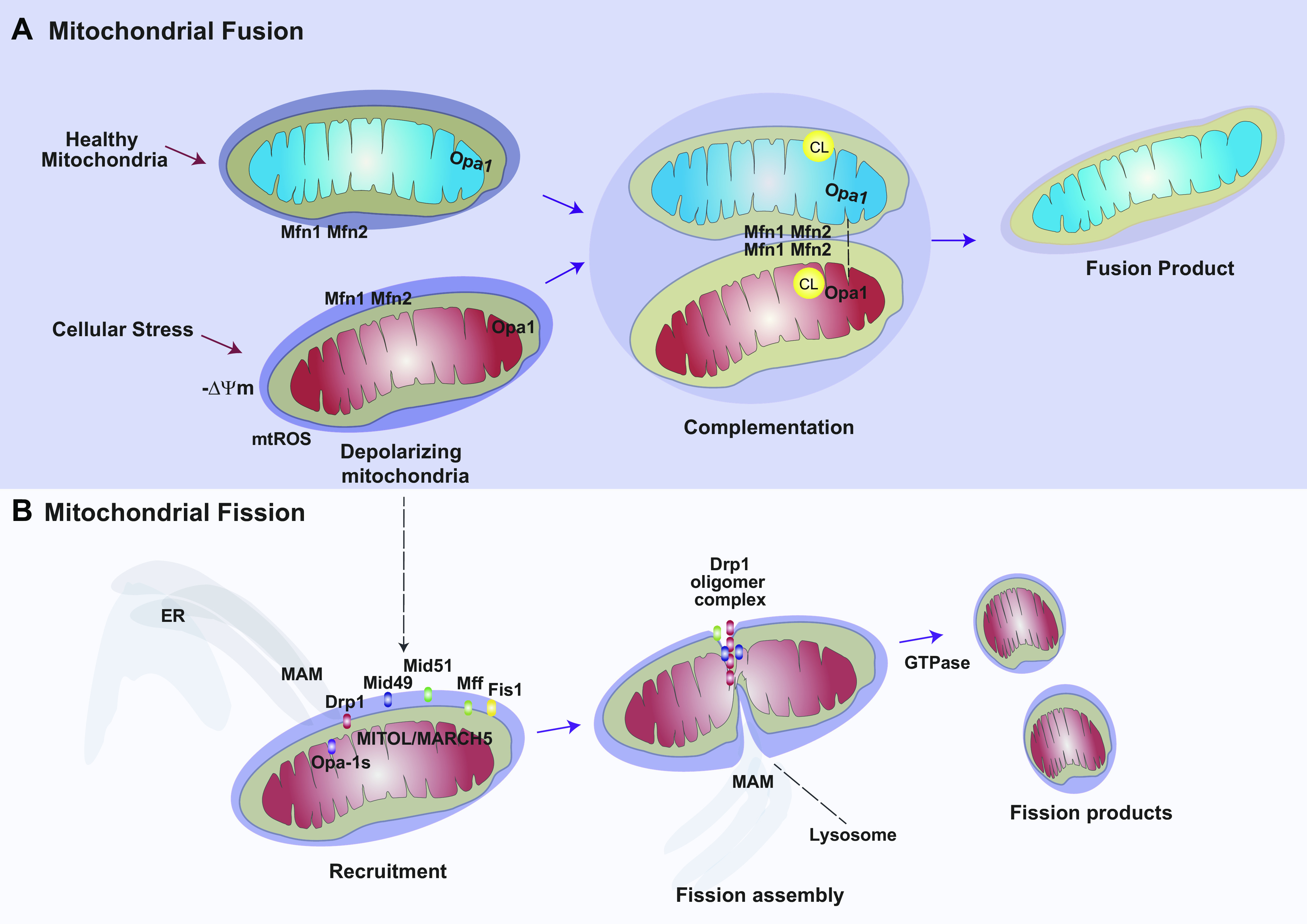
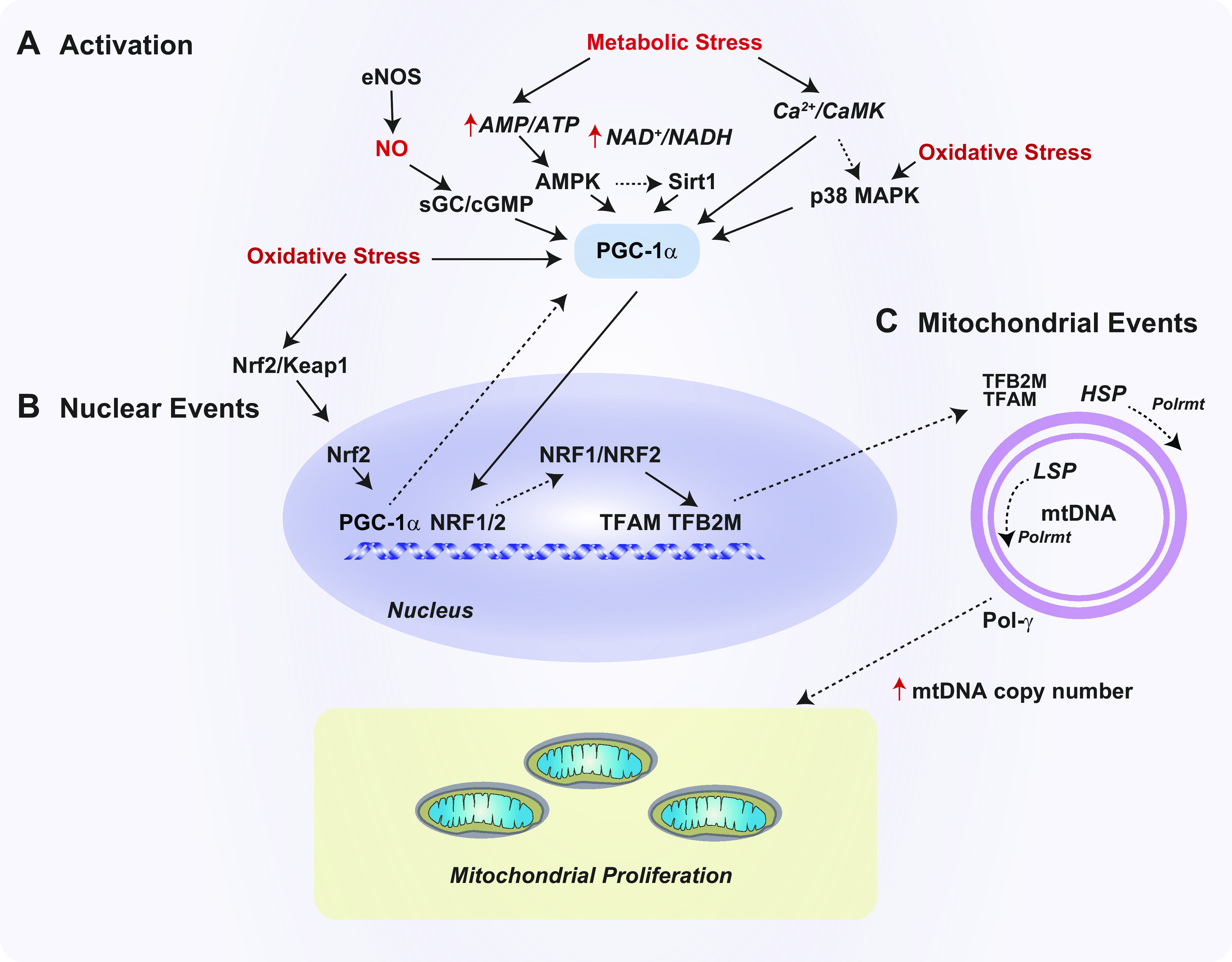
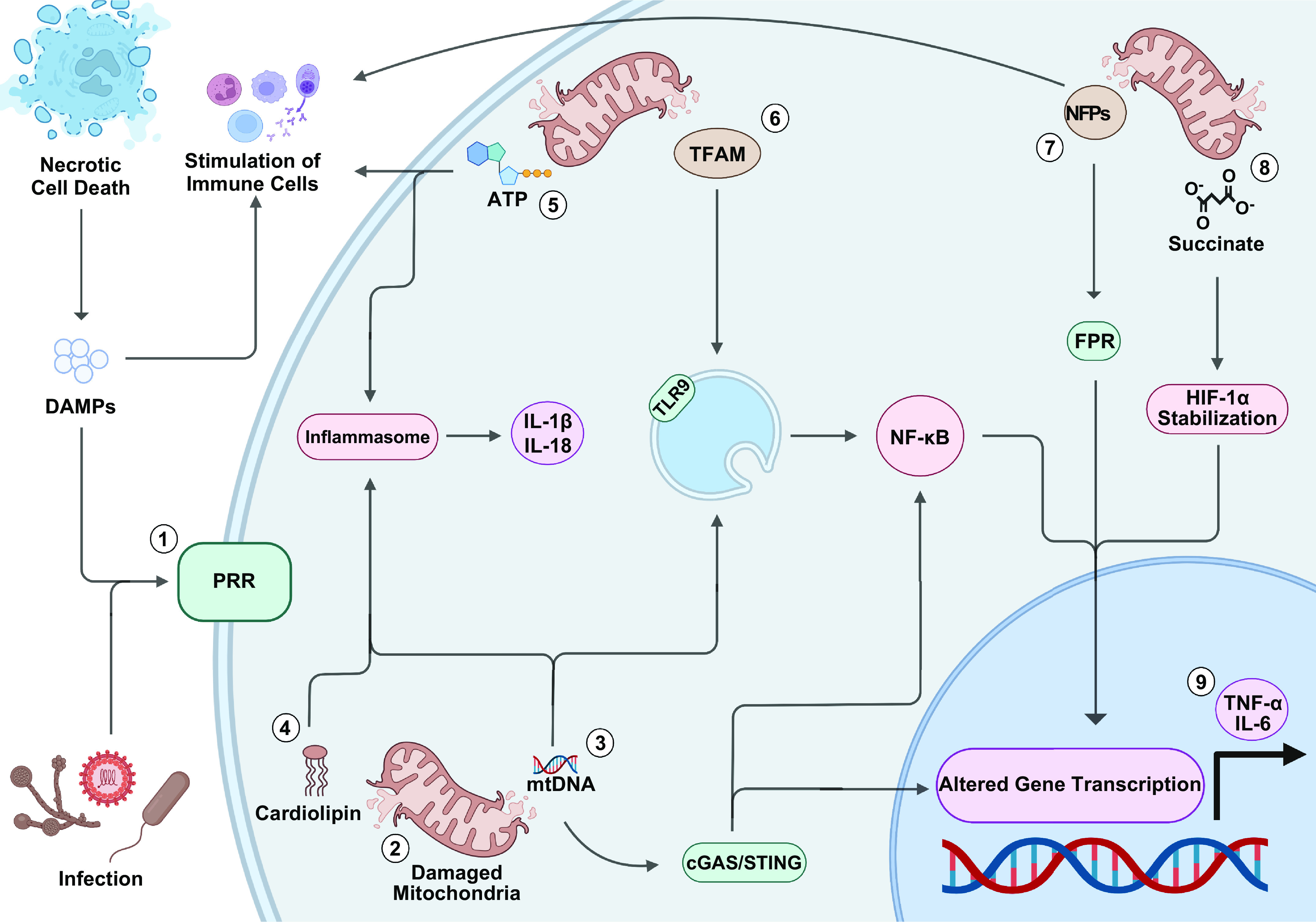
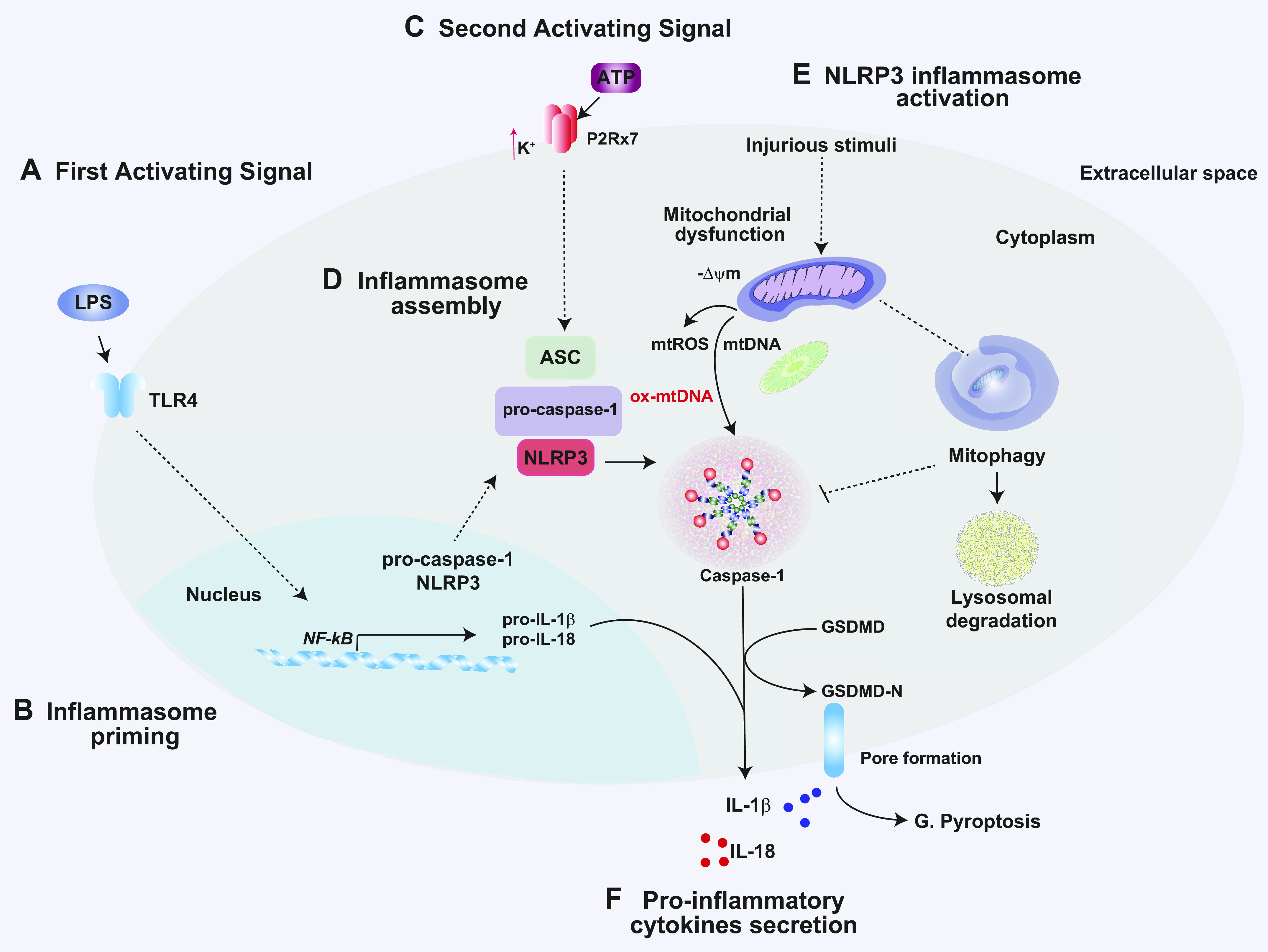
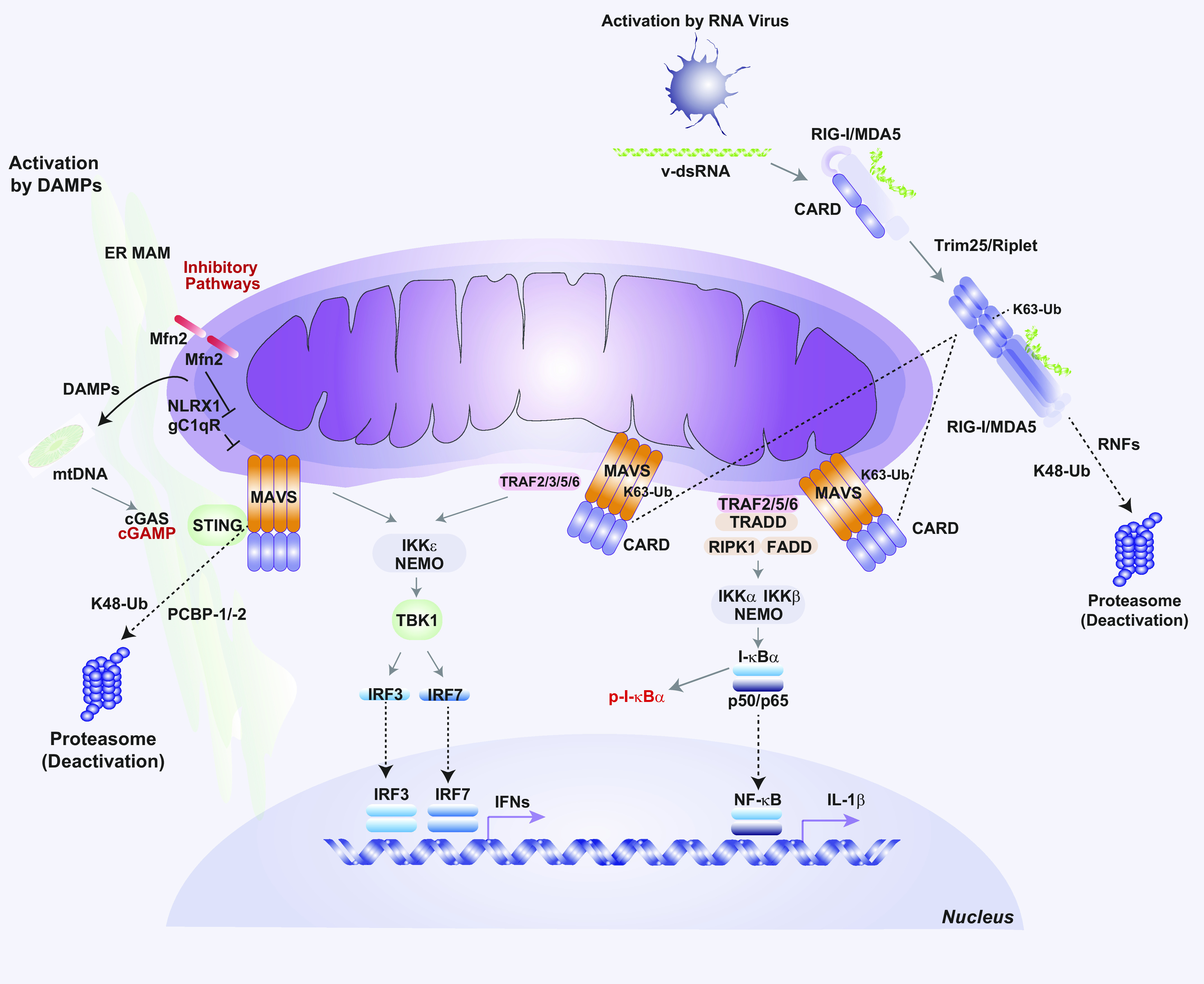
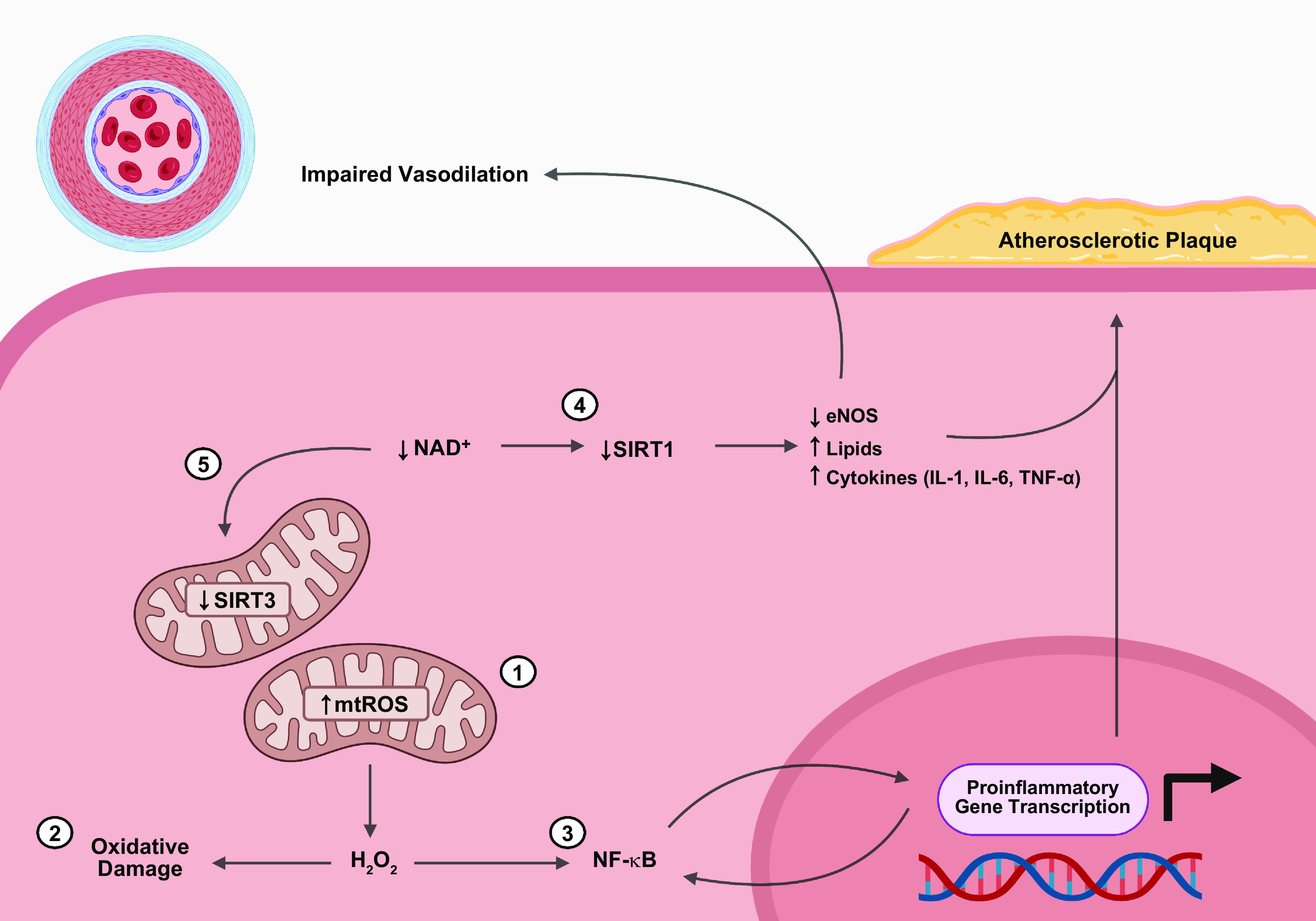
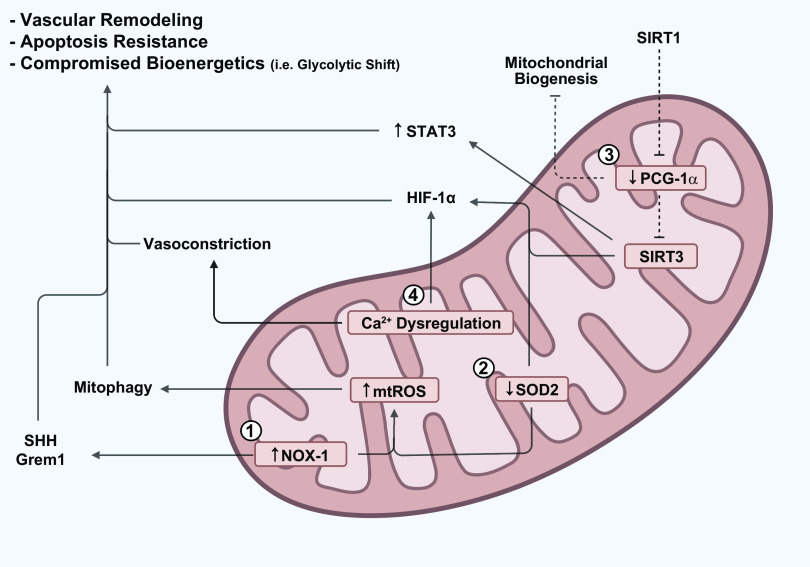
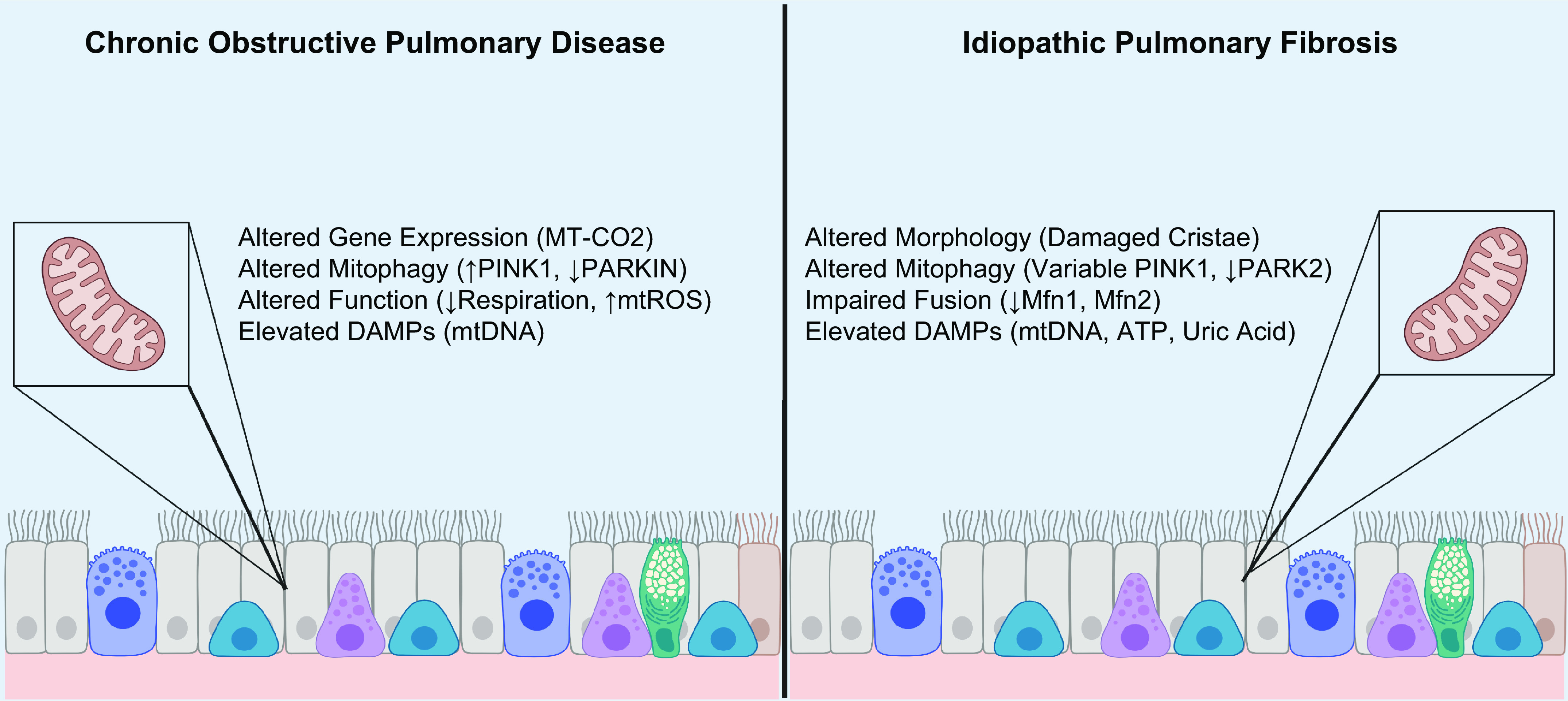
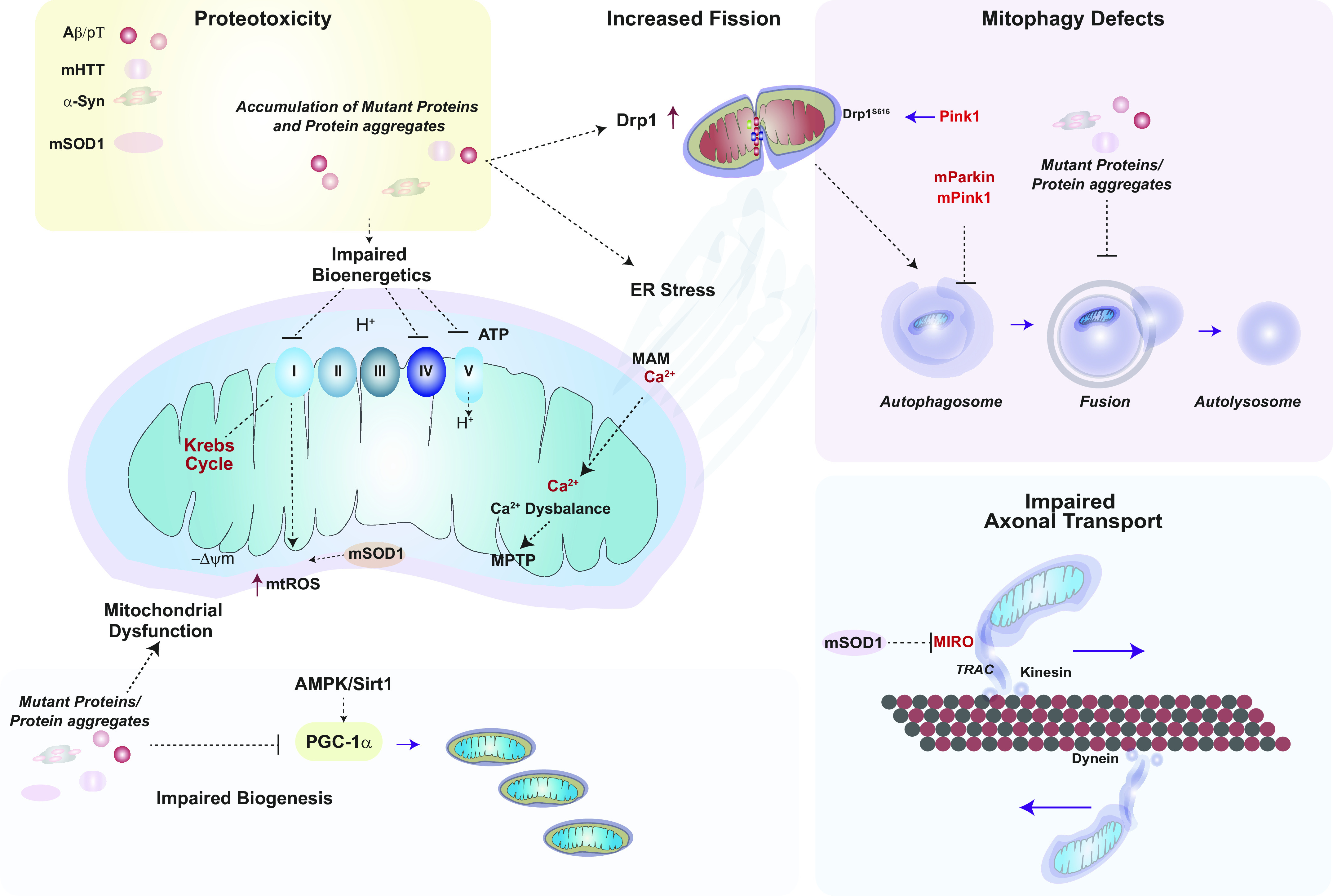
Mitochondria are well known as organelles responsible for the maintenance of cellular bioenergetics through the production of ATP. Although oxidative phosphorylation may be their most important function, mitochondria are also integral for the synthesis of metabolic precursors, calcium regulation, the production of reactive oxygen species, immune signaling, and apoptosis. Considering the breadth of their responsibilities, mitochondria are fundamental for cellular metabolism and homeostasis. Appreciating this significance, translational medicine has begun to investigate how mitochondrial dysfunction can represent a harbinger of disease. In this review, we provide a detailed overview of mitochondrial metabolism, cellular bioenergetics, mitochondrial dynamics, autophagy, mitochondrial damage-associated molecular patterns, mitochondria-mediated cell death pathways, and how mitochondrial dysfunction at any of these levels is associated with disease pathogenesis. Mitochondria-dependent pathways may thereby represent an attractive therapeutic target for ameliorating human disease.
Keywords: inflammation; mitochondria; mitochondrial dynamics; mitochondrial dysfunction; mitophagy.
Conflict of interest statement
S.W.R. is a current employee and stockholder of Proterris Inc. and a former Weill Cornell employee. A.M.K.C. is a cofounder and equity stockholder of Proterris, which develops therapeutic uses for carbon monoxide. A.M.K.C. has a use patent on CO. Additionally, A.M.K.C. has a patent in COPD. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical