

Quantifying Shared and Unique Gene Content across 17 Microbial Ecosystems
- PMID: 37022232
- PMCID: PMC10134805
- DOI: 10.1128/msystems.00118-23
Quantifying Shared and Unique Gene Content across 17 Microbial Ecosystems
Abstract
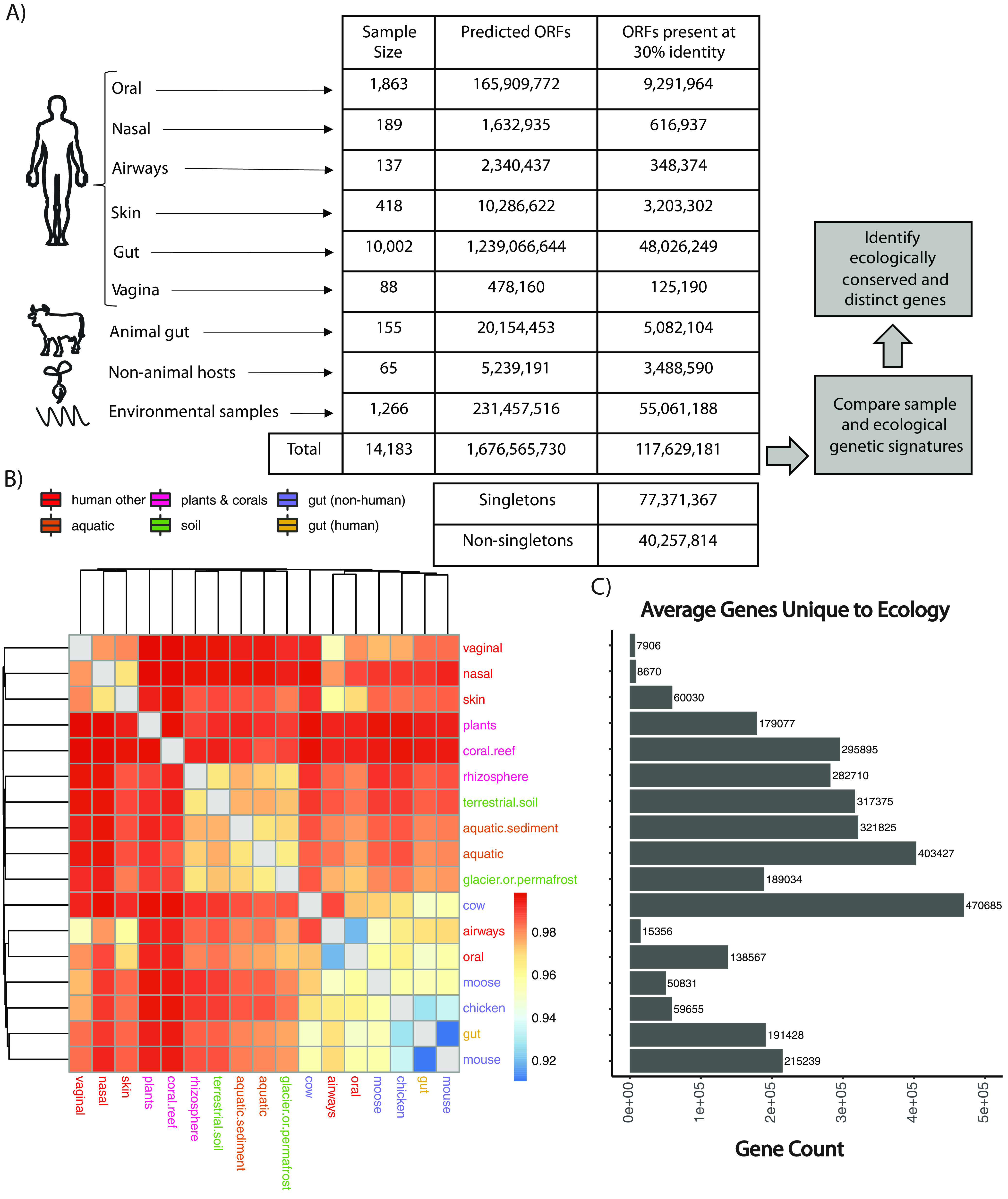
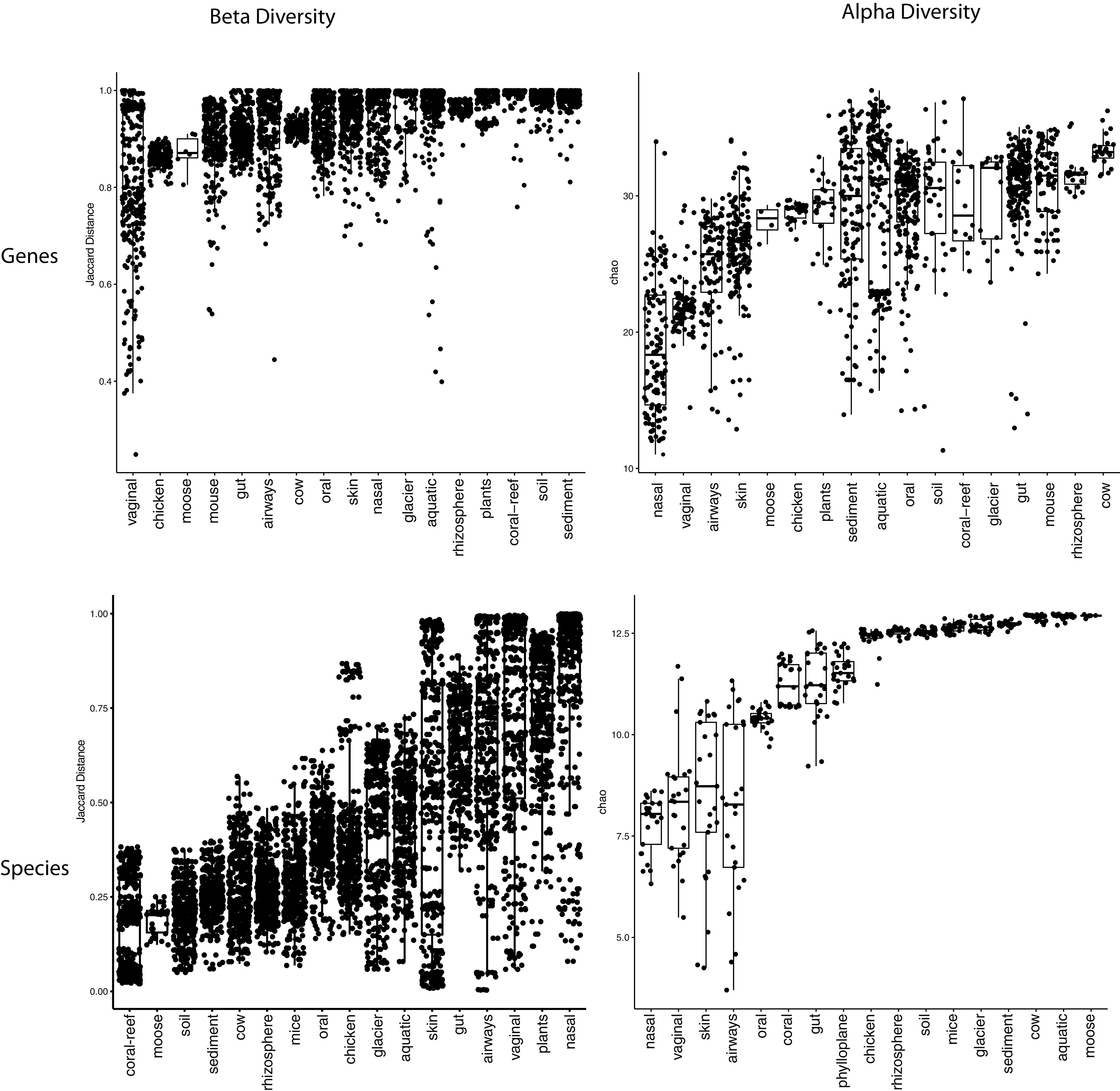
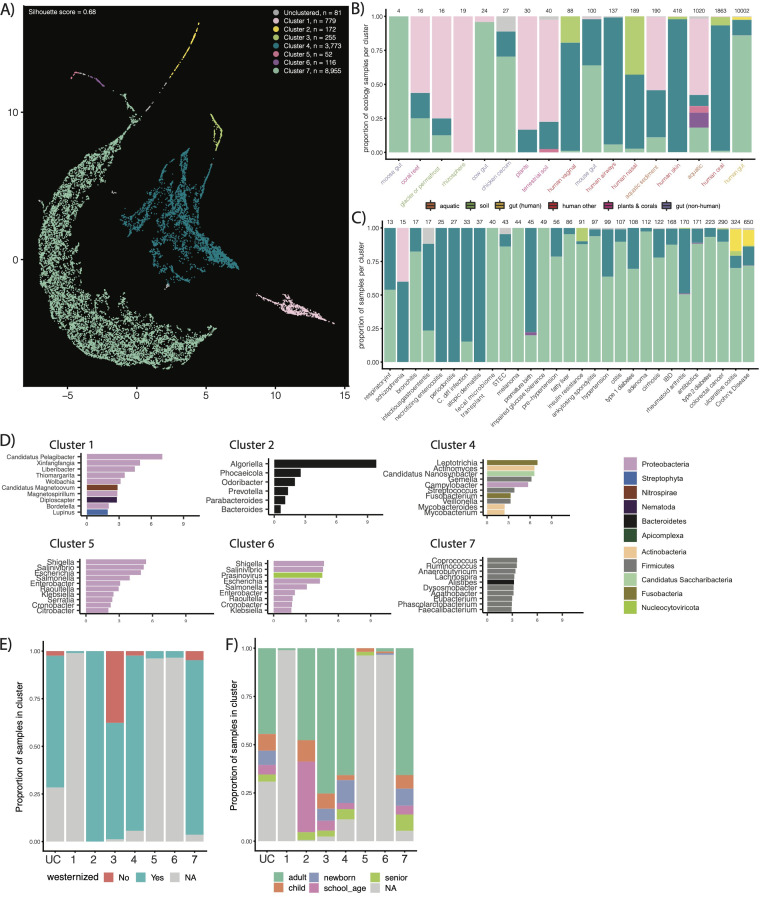
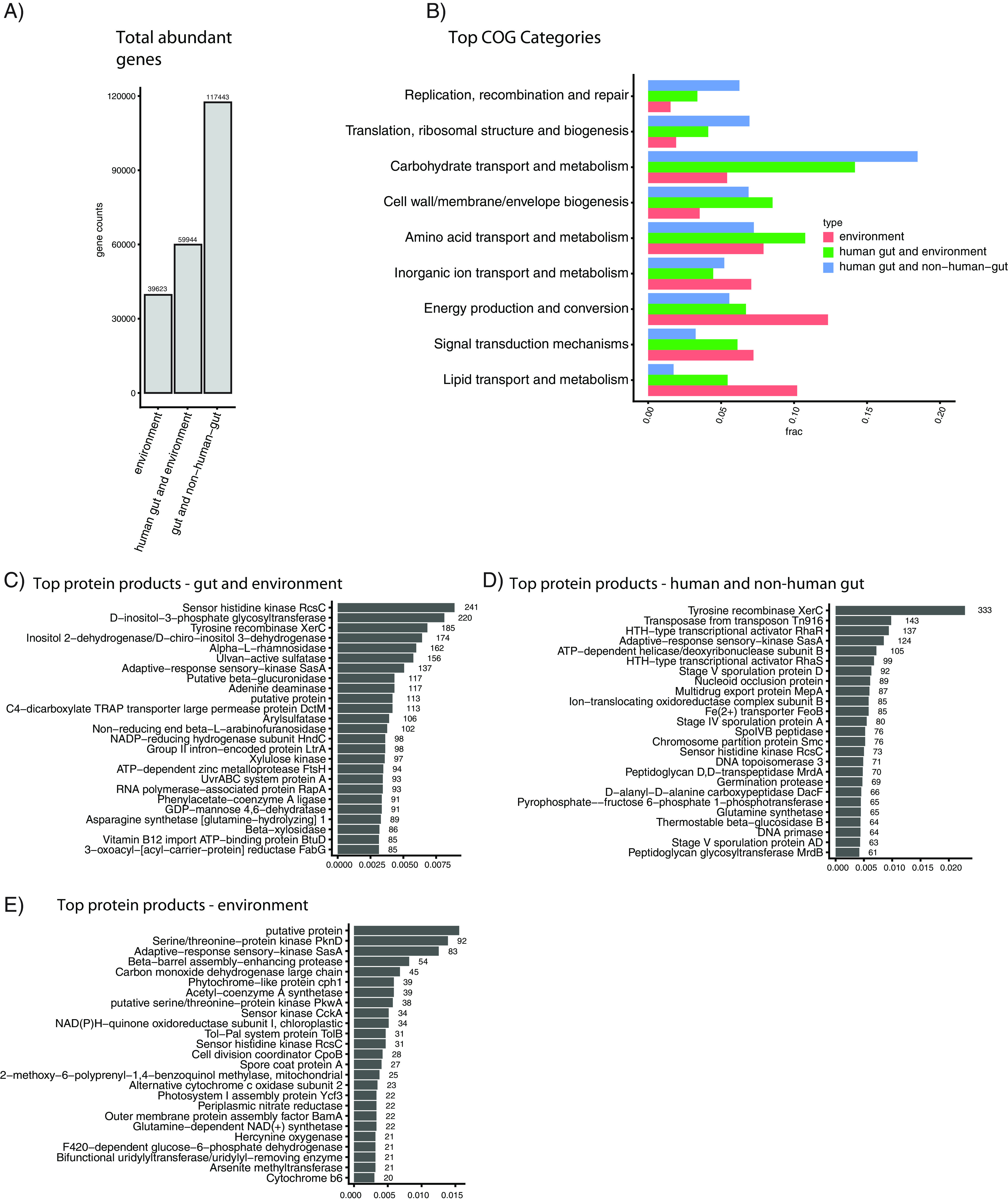
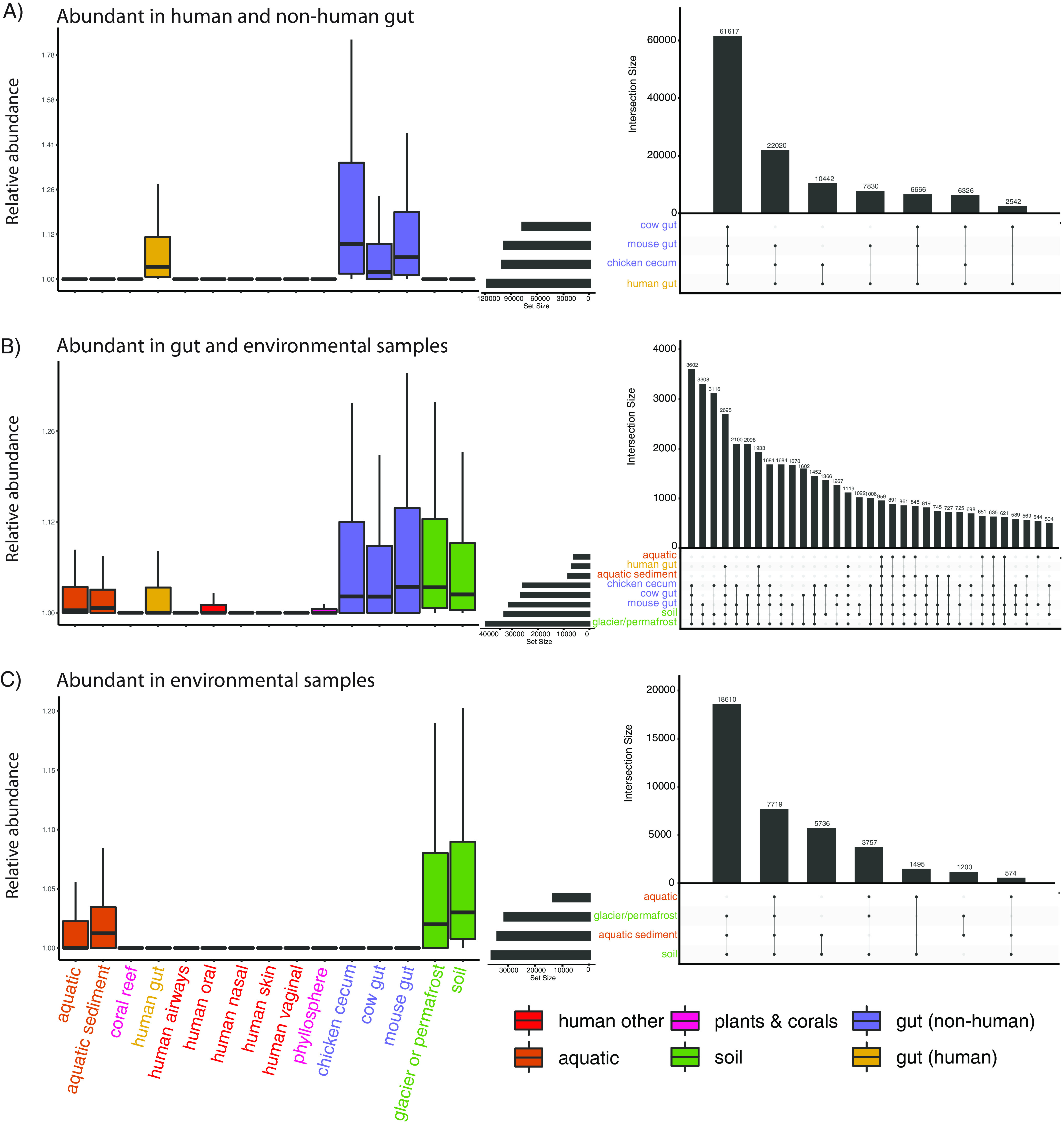
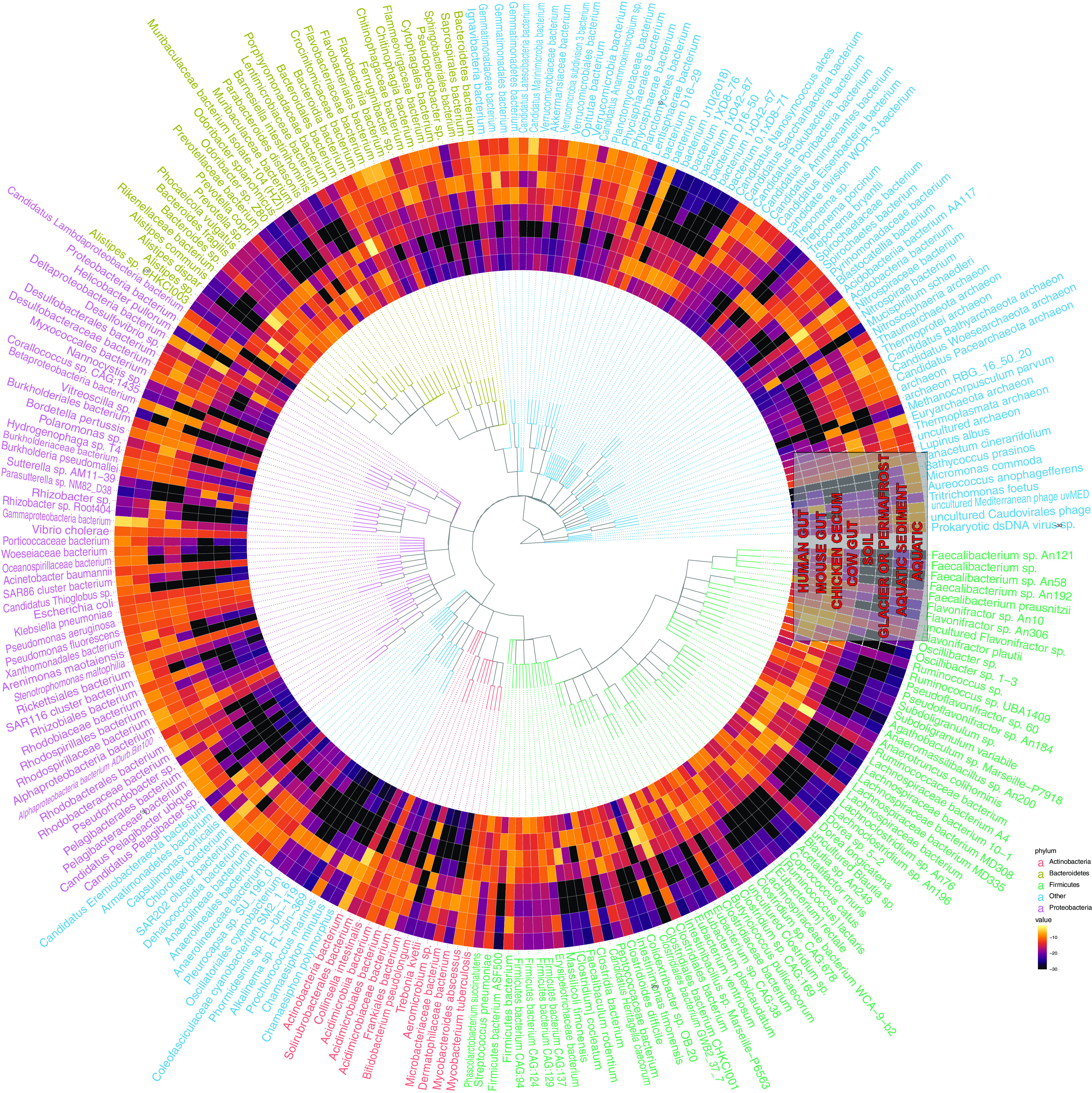
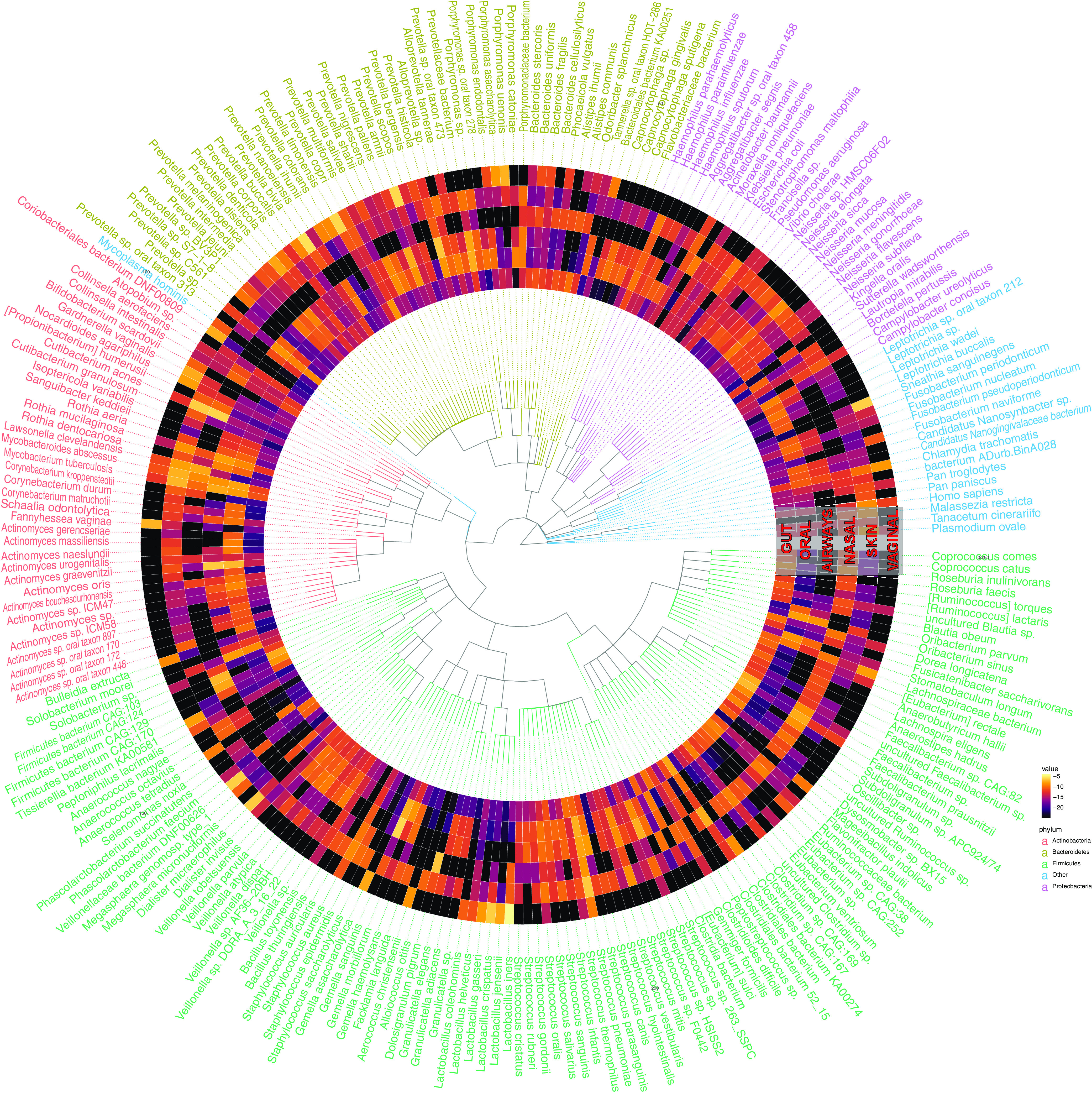
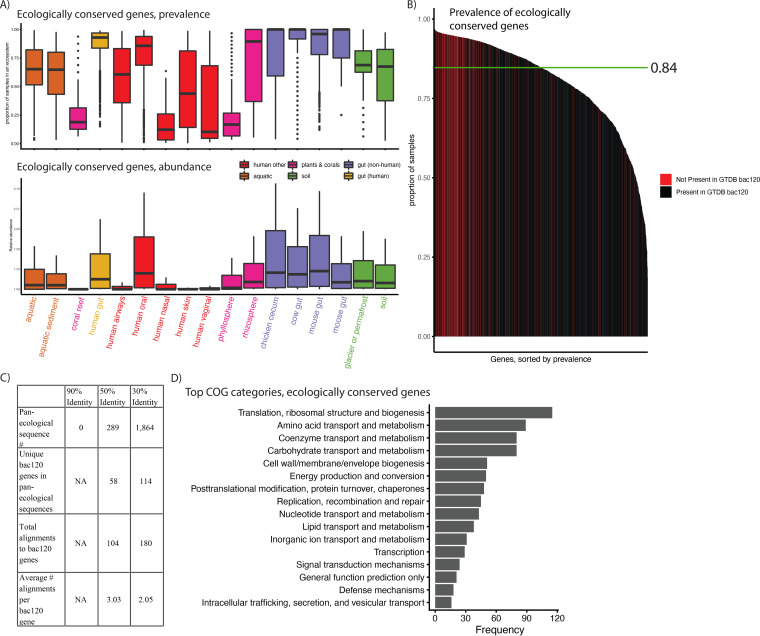
Measuring microbial diversity is traditionally based on microbe taxonomy. Here, in contrast, we aimed to quantify heterogeneity in microbial gene content across 14,183 metagenomic samples spanning 17 ecologies, including 6 human associated, 7 nonhuman host associated, and 4 in other nonhuman host environments. In total, we identified 117,629,181 nonredundant genes. The vast majority of genes (66%) occurred in only one sample (i.e., "singletons"). In contrast, we found 1,864 sequences present in every metagenome, but not necessarily every bacterial genome. Additionally, we report data sets of other ecology-associated genes (e.g., abundant in only gut ecosystems) and simultaneously demonstrated that prior microbiome gene catalogs are both incomplete and inaccurately cluster microbial genetic life (e.g., at gene sequence identities that are too restrictive). We provide our results and the sets of environmentally differentiating genes described above at http://www.microbial-genes.bio. IMPORTANCE The amount of shared genetic elements has not been quantified between the human microbiome and other host- and non-host-associated microbiomes. Here, we made a gene catalog of 17 different microbial ecosystems and compared them. We show that most species shared between environment and human gut microbiomes are pathogens and that prior gene catalogs described as "nearly complete" are far from it. Additionally, over two-thirds of all genes only appear in a single sample, and only 1,864 genes (0.001%) are found in all types of metagenomes. These results highlight the large diversity between metagenomes and reveal a new, rare class of genes, those found in every type of metagenome, but not every microbial genome.
Keywords: bioinformatics; human microbiome; metagenomics.
Conflict of interest statement
The authors declare a conflict of interest. Aleksandar D. Kostic is an advisor at FitBiomics. Chirag J. Patel is a cofounder of XY.ai. Braden T. Tierney consults for Seed Health on microbiome study design and analysis.
Figures
References
-
- Knights D, Silverberg MS, Weersma RK, Gevers D, Dijkstra G, Huang H, Tyler AD, van Sommeren S, Imhann F, Stempak JM, Huang H, Vangay P, Al-Ghalith GA, Russell C, Sauk J, Knight J, Daly MJ, Huttenhower C, Xavier RJ. 2014. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med 6:107. doi: 10.1186/s13073-014-0107-1. - DOI - PMC - PubMed
-
- Le Goallec A, Tierney BT, Luber JM, Cofer EM, Kostic AD, Patel CJ. 2020. A systematic machine learning and data type comparison yields metagenomic predictors of infant age, sex, breastfeeding, antibiotic usage, country of origin, and delivery type. PLoS Comput Biol 16:e1007895. doi: 10.1371/journal.pcbi.1007895. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
