

Treatment effects of soluble guanylate cyclase modulation on diabetic kidney disease at single-cell resolution
- PMID: 37023747
- PMCID: PMC10140477
- DOI: 10.1016/j.xcrm.2023.100992
Treatment effects of soluble guanylate cyclase modulation on diabetic kidney disease at single-cell resolution
Abstract
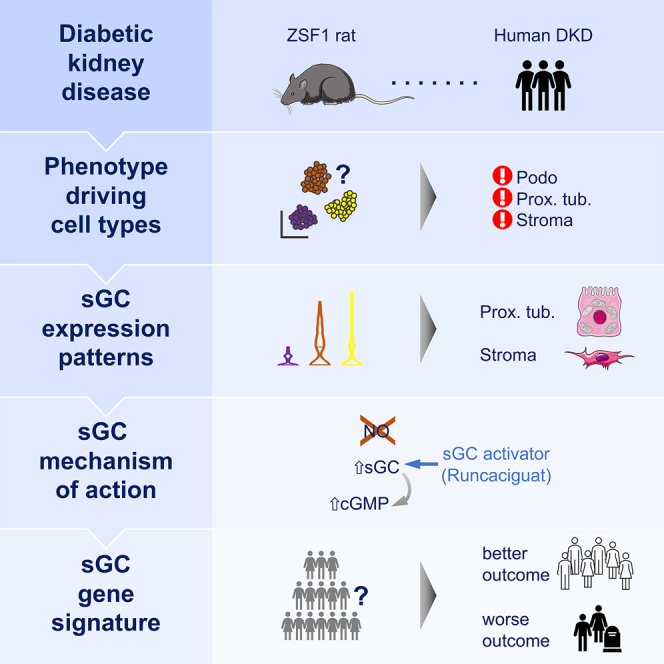
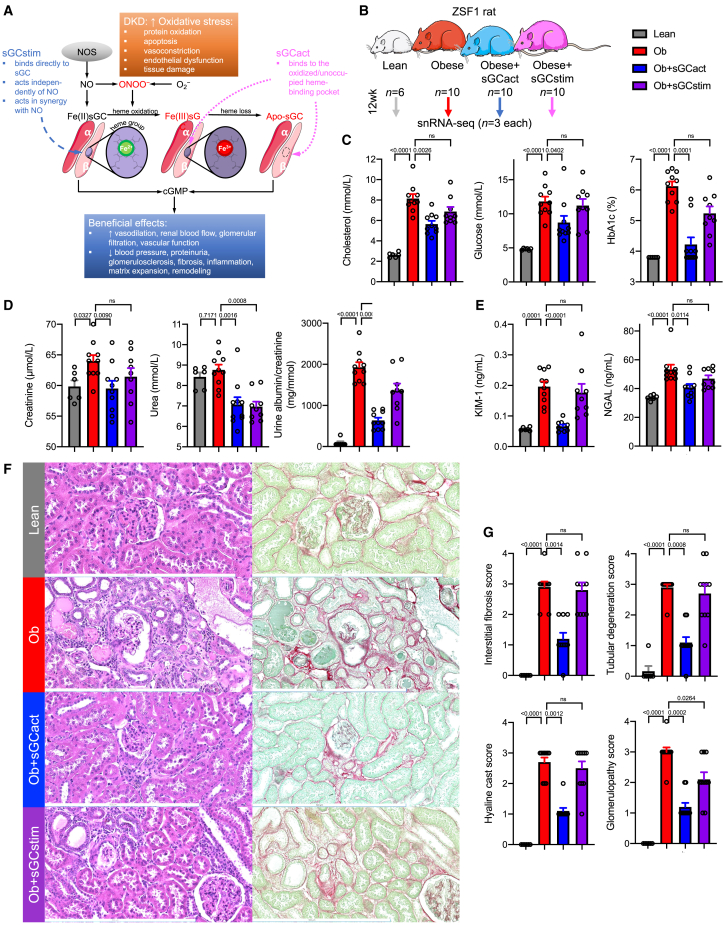
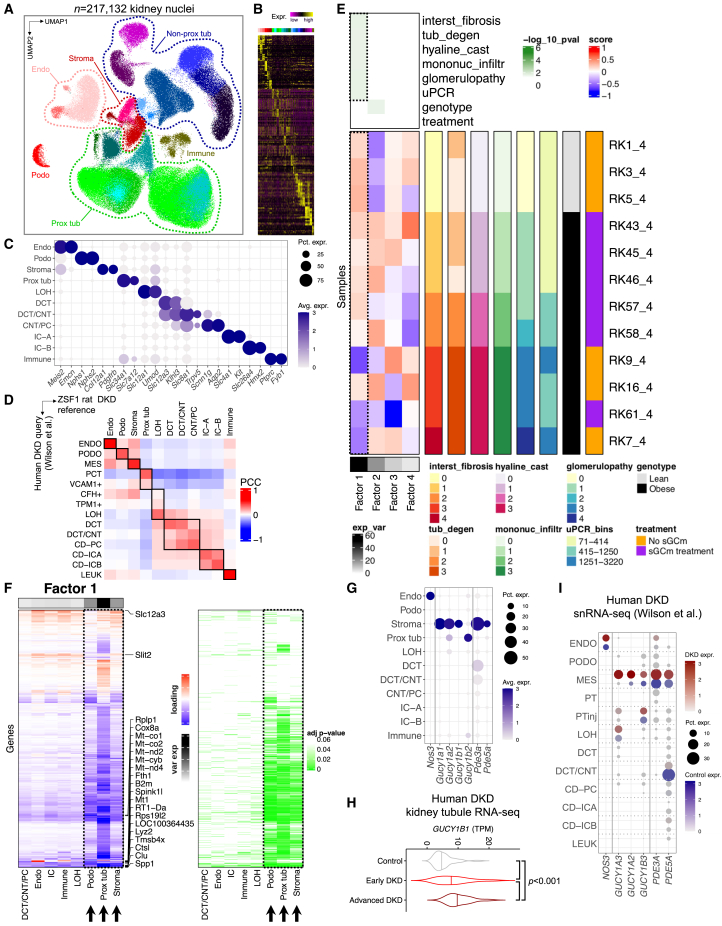
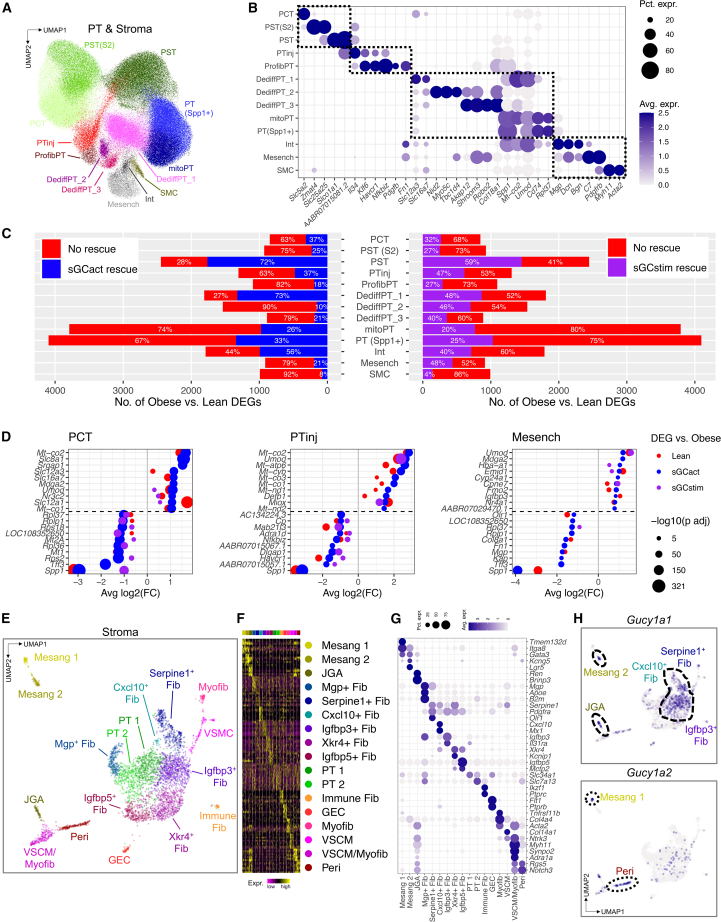
Diabetic kidney disease (DKD) is the most common cause of renal failure. Therapeutics development is hampered by our incomplete understanding of animal models on a cellular level. We show that ZSF1 rats recapitulate human DKD on a phenotypic and transcriptomic level. Tensor decomposition prioritizes proximal tubule (PT) and stroma as phenotype-relevant cell types exhibiting a continuous lineage relationship. As DKD features endothelial dysfunction, oxidative stress, and nitric oxide depletion, soluble guanylate cyclase (sGC) is a promising DKD drug target. sGC expression is specifically enriched in PT and stroma. In ZSF1 rats, pharmacological sGC activation confers considerable benefits over stimulation and is mechanistically related to improved oxidative stress regulation, resulting in enhanced downstream cGMP effects. Finally, we define sGC gene co-expression modules, which allow stratification of human kidney samples by DKD prevalence and disease-relevant measures such as kidney function, proteinuria, and fibrosis, underscoring the relevance of the sGC pathway to patients.
Keywords: CDK; DKD; NO; ZSF1 rat; chronic kidney disease; diabetic kidney disease; gene-regulatory network; nitric oxide; oxidative stress; sGC; single-cell RNA-seq; soluble guanylate cyclase; tensor decomposition; weighted gene correlation network analysis.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.F., J.V., I.M., K. Siudak, F.E., P.S., M.G., and M.P. are employees of Bayer AG. M.S.B. reports consultancy: Boehringer Ingelheim; editorial board membership: Journal of the American Society of Nephrology. K. Susztak reports research support: AstraZeneca, Bayer, Boehringer Ingelheim, Calico, Genentech, Gilead, GSK, Jnana, Lilly, Maze, Merck, Novartis, Novo Nordisk, Regeneron, Variant Bio, and Ventus; advisory board membership: Jnana Therapeutics and Pfizer; consultancy: AstraZeneca, Bayer, GSK, Jnana Therapeutics, Maze, Novo Nordisk, Pfizer, and Ventus; patents: Jag1- and Notch-based targeting of chronic kidney disease; editorial board membership: Cell Metabolism, eBioMedicine, Journal of the American Society of Nephrology, Journal of Clinical Investigation, Kidney International, and Med.
Figures
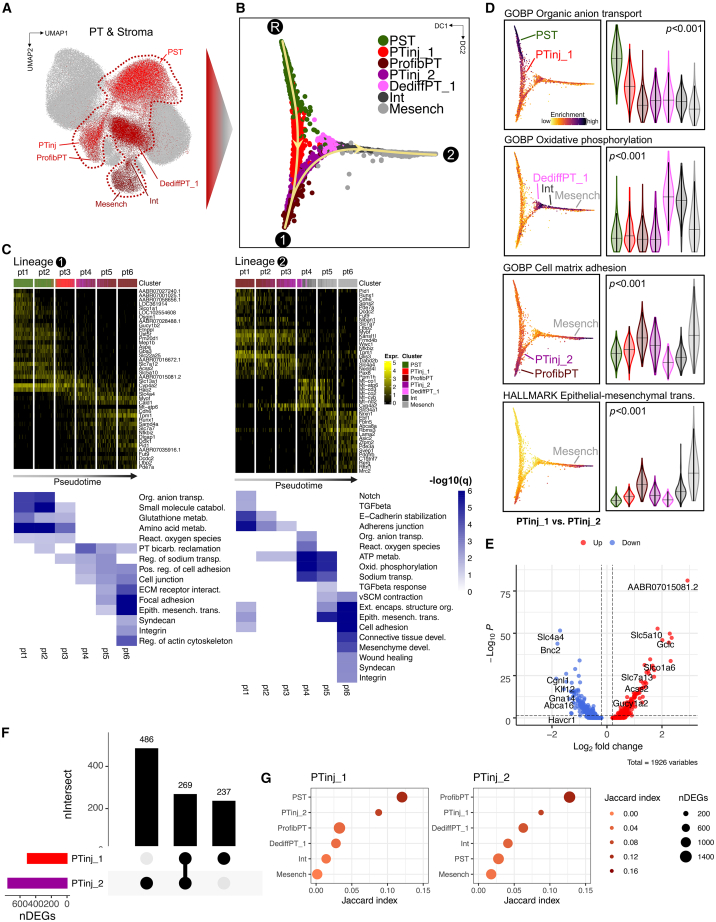
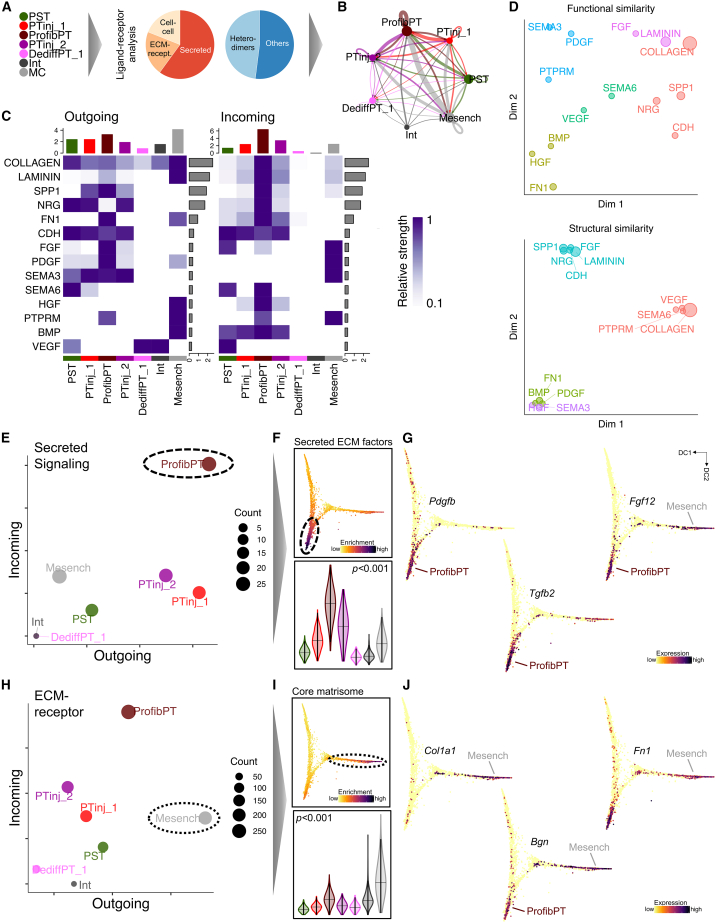
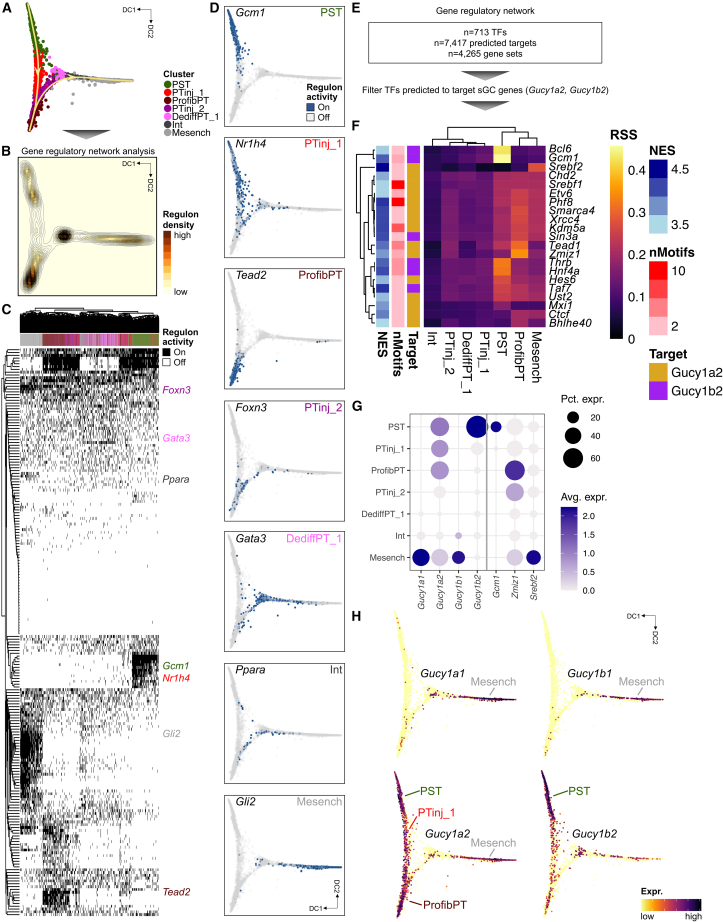
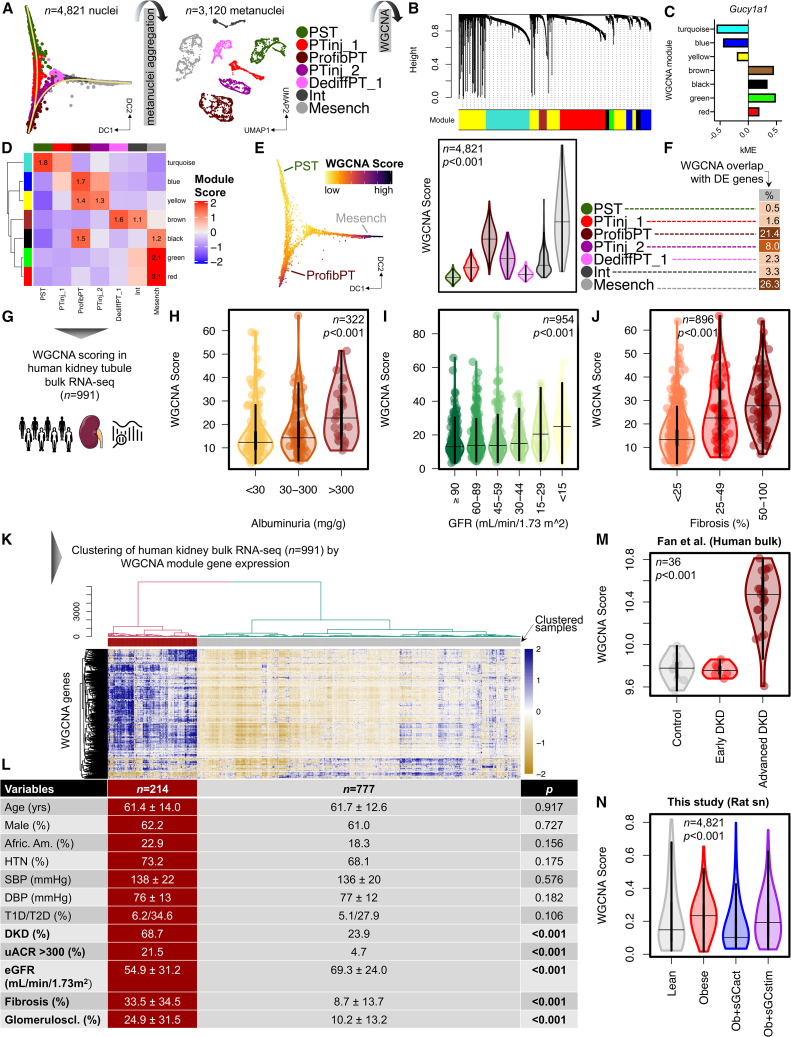
References
-
- U.S. Renal Data System . National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD: 2014. USRDS 2014 Annual Data Report: Atlas of End-Stage Renal Disease in the United States, National Institutes of Health.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
