

Precise modulation of transcription factor levels identifies features underlying dosage sensitivity
- PMID: 37024583
- PMCID: PMC10181932
- DOI: 10.1038/s41588-023-01366-2
Precise modulation of transcription factor levels identifies features underlying dosage sensitivity
Abstract
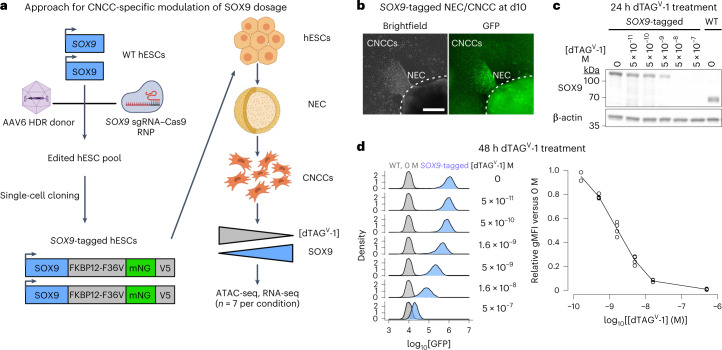
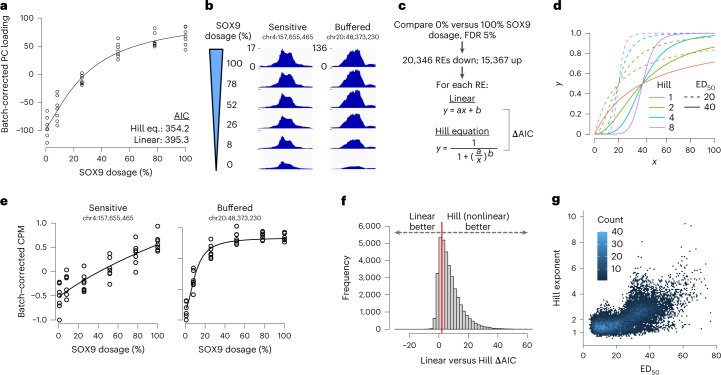
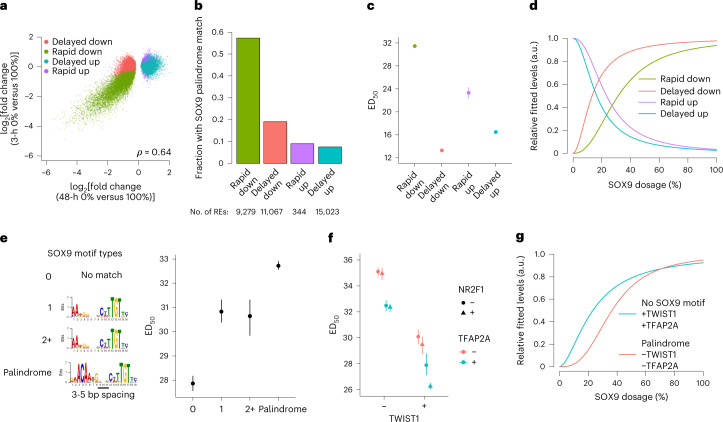
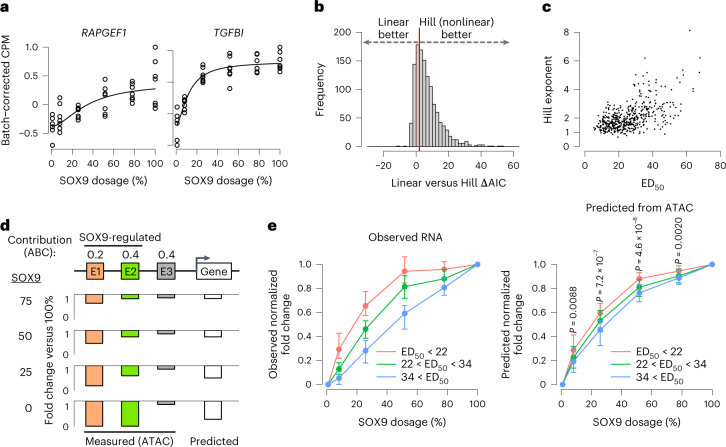
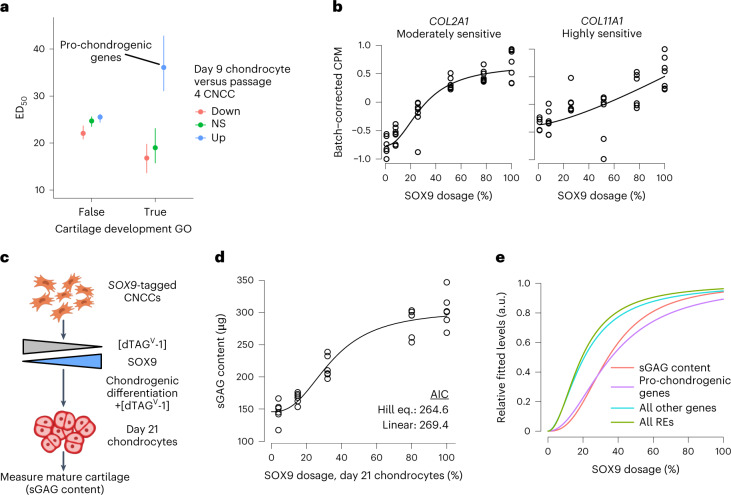
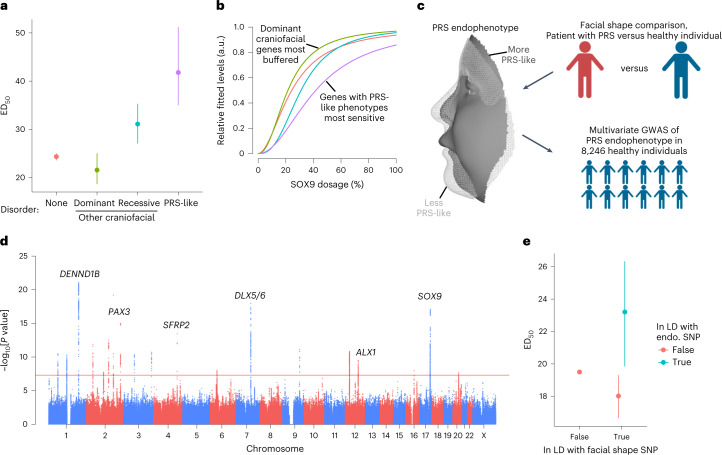
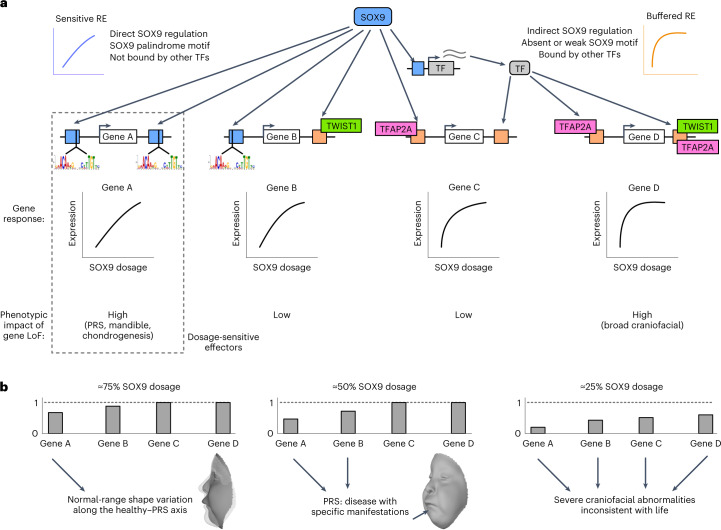
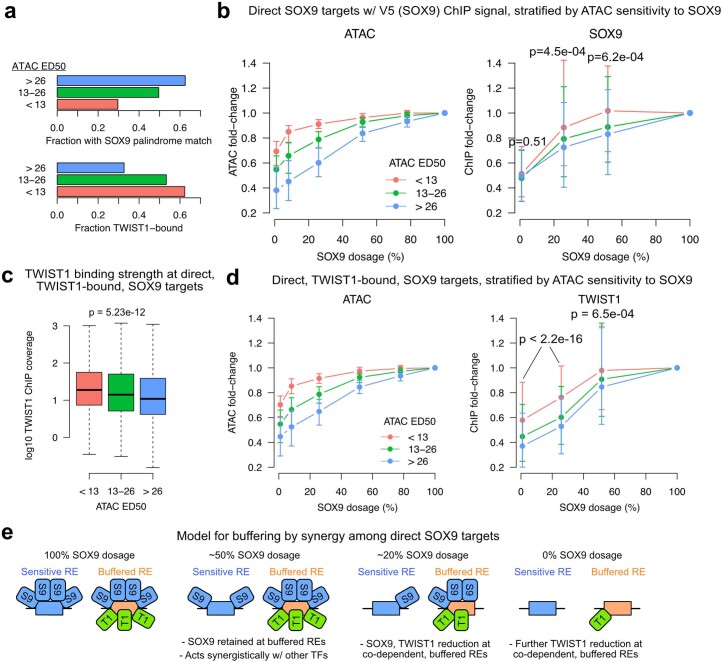
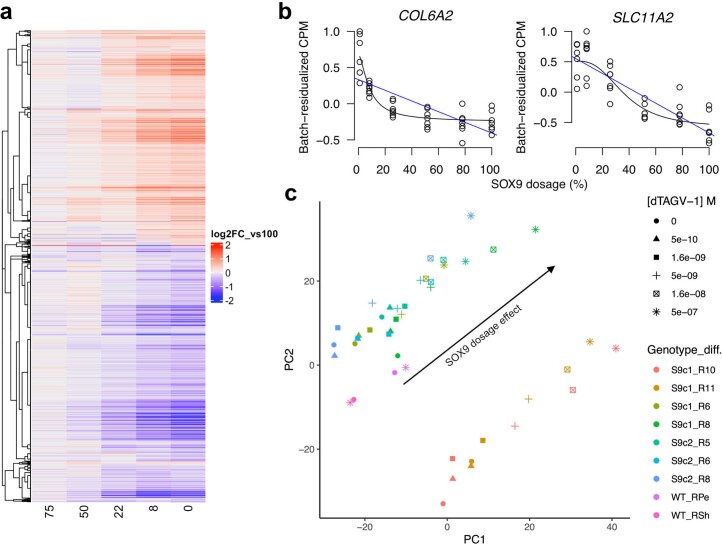
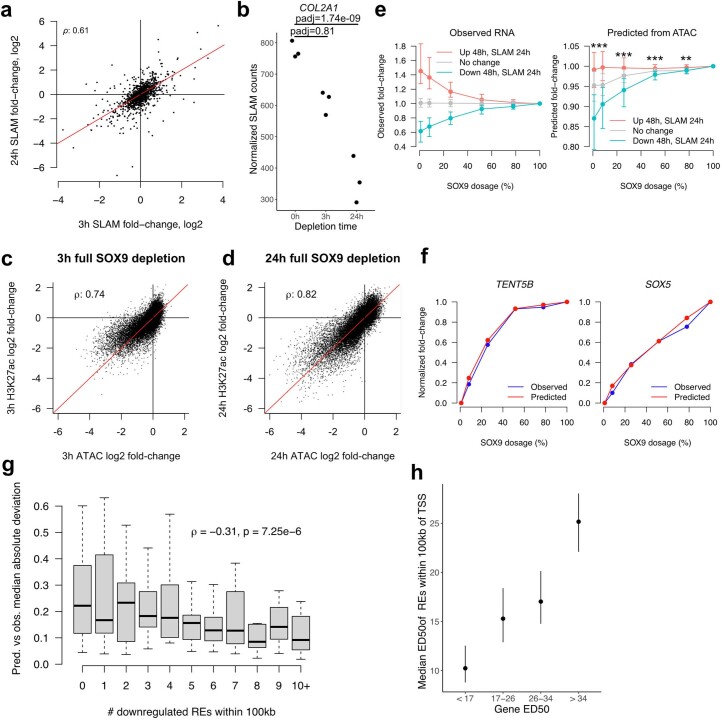
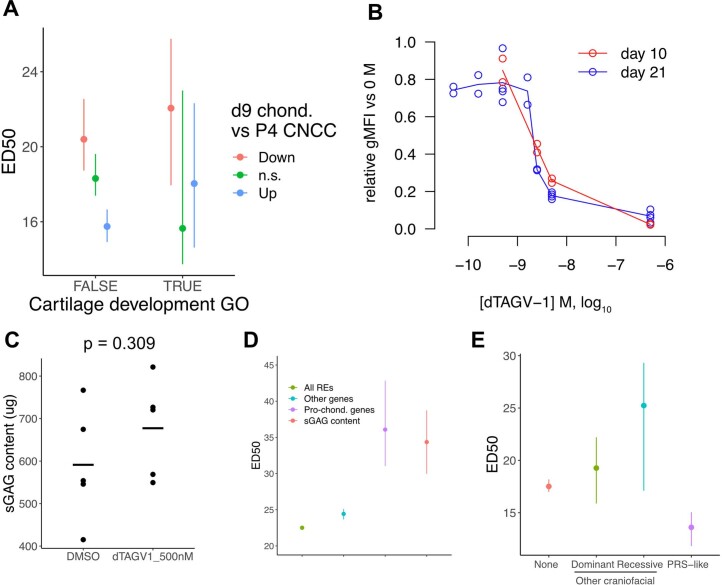
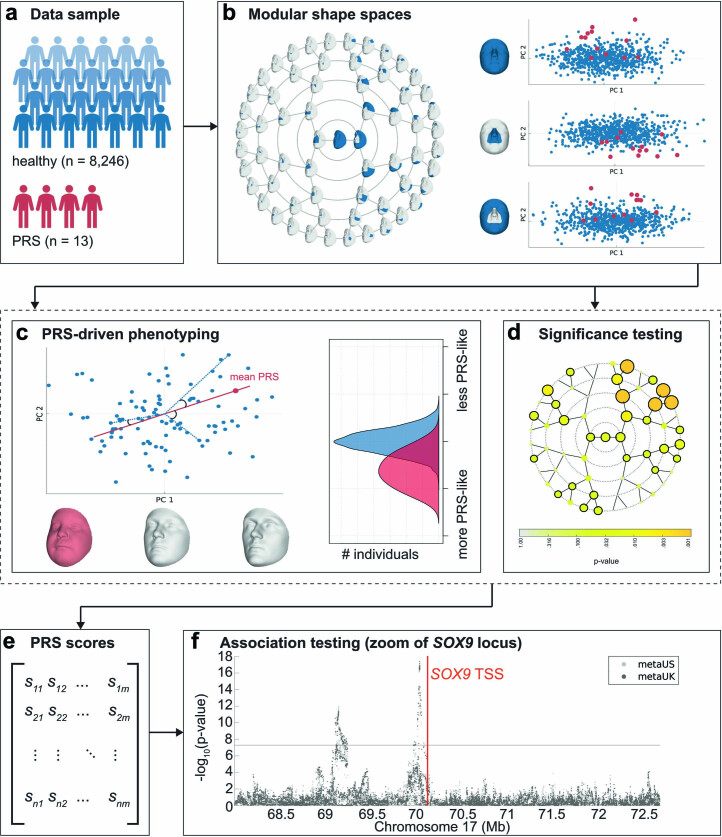
Transcriptional regulation exhibits extensive robustness, but human genetics indicates sensitivity to transcription factor (TF) dosage. Reconciling such observations requires quantitative studies of TF dosage effects at trait-relevant ranges, largely lacking so far. TFs play central roles in both normal-range and disease-associated variation in craniofacial morphology; we therefore developed an approach to precisely modulate TF levels in human facial progenitor cells and applied it to SOX9, a TF associated with craniofacial variation and disease (Pierre Robin sequence (PRS)). Most SOX9-dependent regulatory elements (REs) are buffered against small decreases in SOX9 dosage, but REs directly and primarily regulated by SOX9 show heightened sensitivity to SOX9 dosage; these RE responses partially predict gene expression responses. Sensitive REs and genes preferentially affect functional chondrogenesis and PRS-like craniofacial shape variation. We propose that such REs and genes underlie the sensitivity of specific phenotypes to TF dosage, while buffering of other genes leads to robust, nonlinear dosage-to-phenotype relationships.
© 2023. The Author(s).
Conflict of interest statement
J.W. serves on the scientific advisory board for Camp4 Therapeutics and Paratus Sciences. All other authors declare no competing interests.
Figures
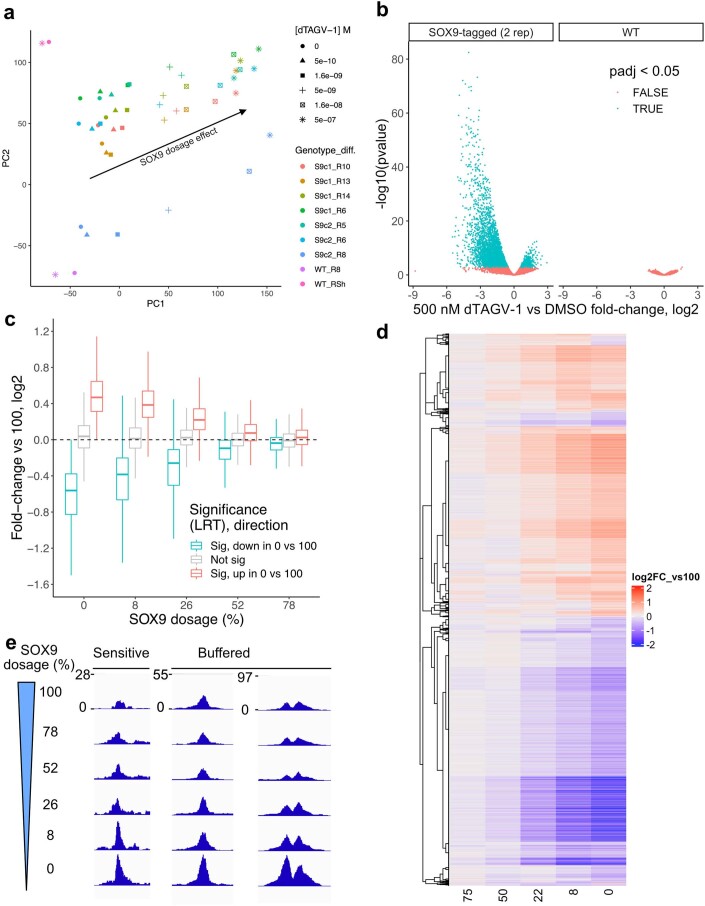
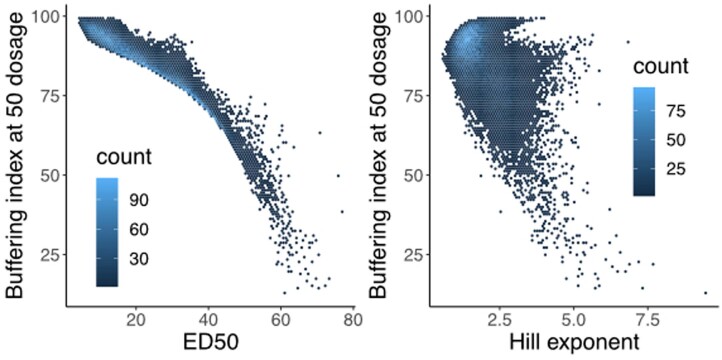
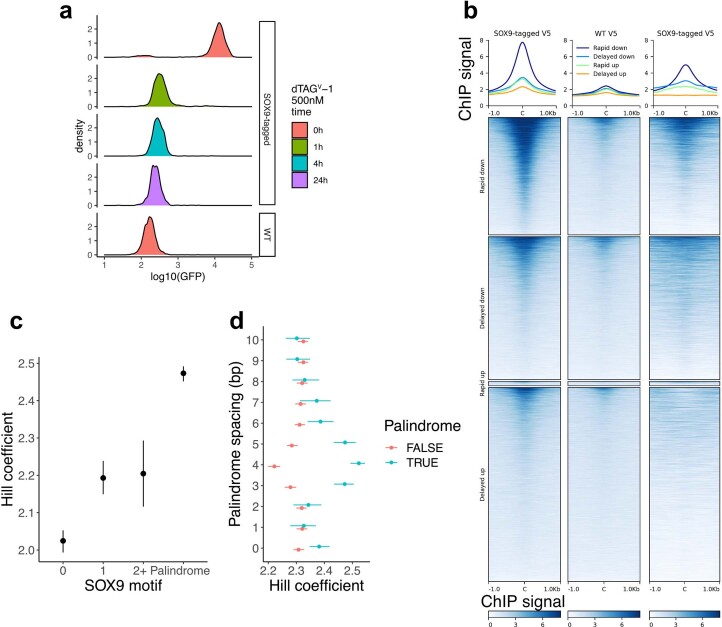
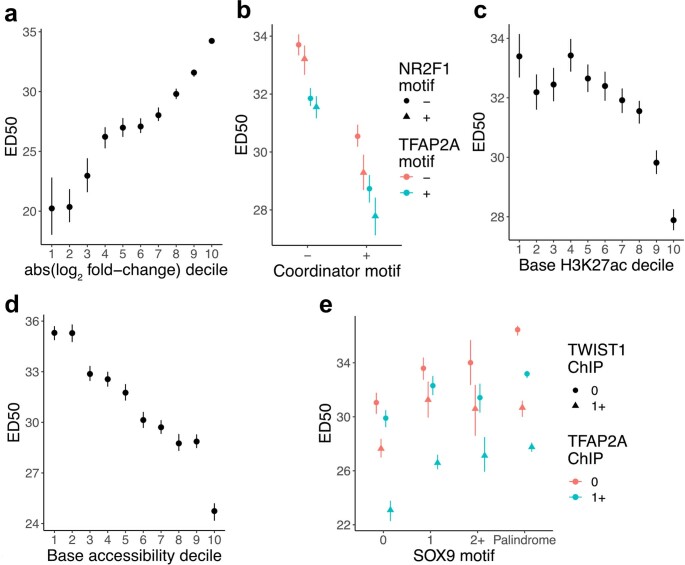
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
