

Quantitative analysis of tRNA abundance and modifications by nanopore RNA sequencing
- PMID: 37024678
- PMCID: PMC10791586
- DOI: 10.1038/s41587-023-01743-6
Quantitative analysis of tRNA abundance and modifications by nanopore RNA sequencing
Abstract
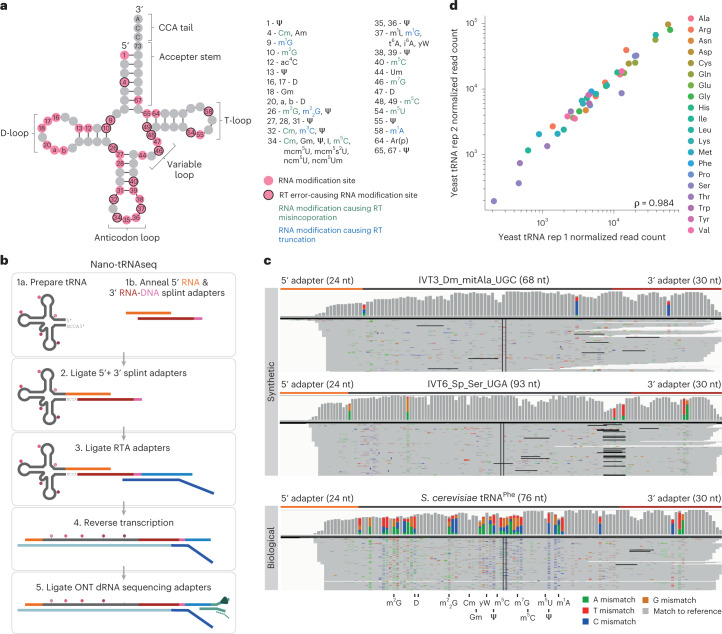
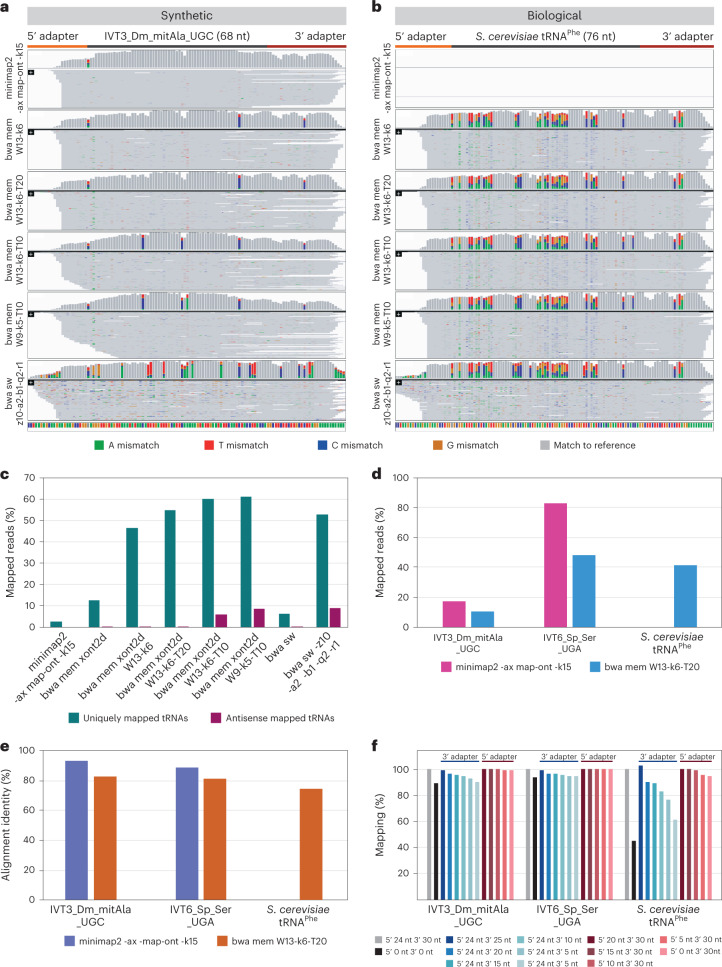
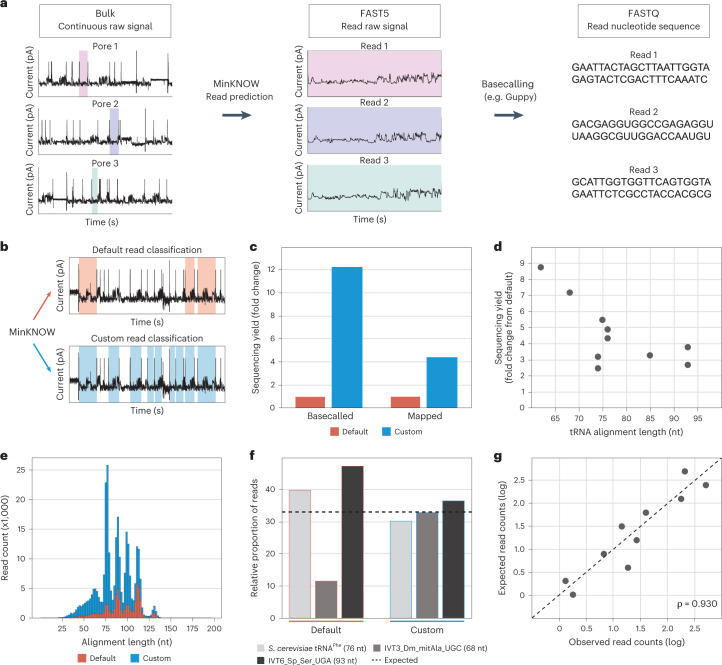
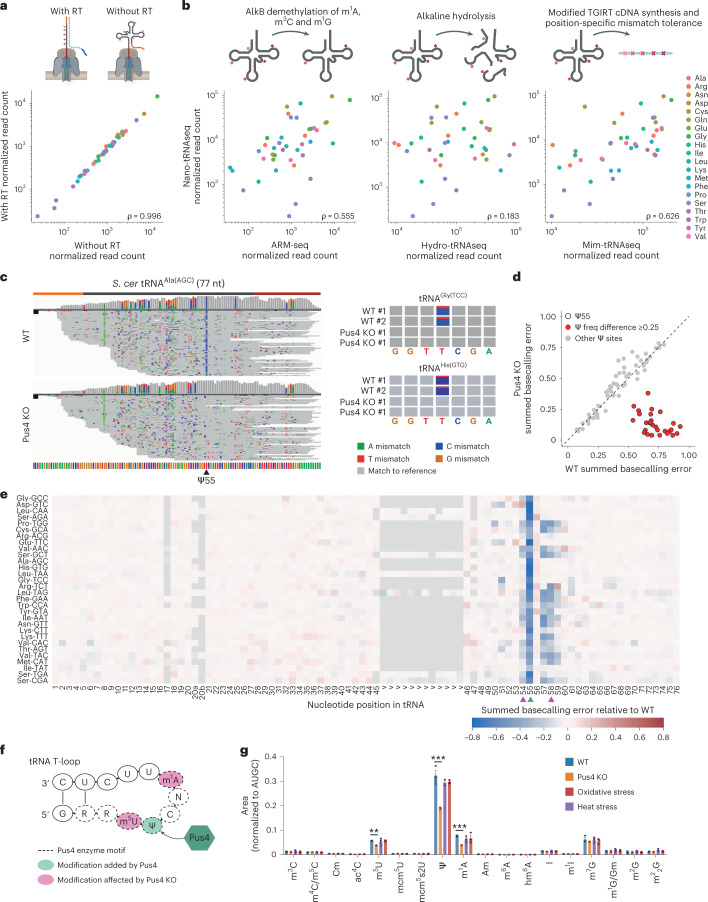
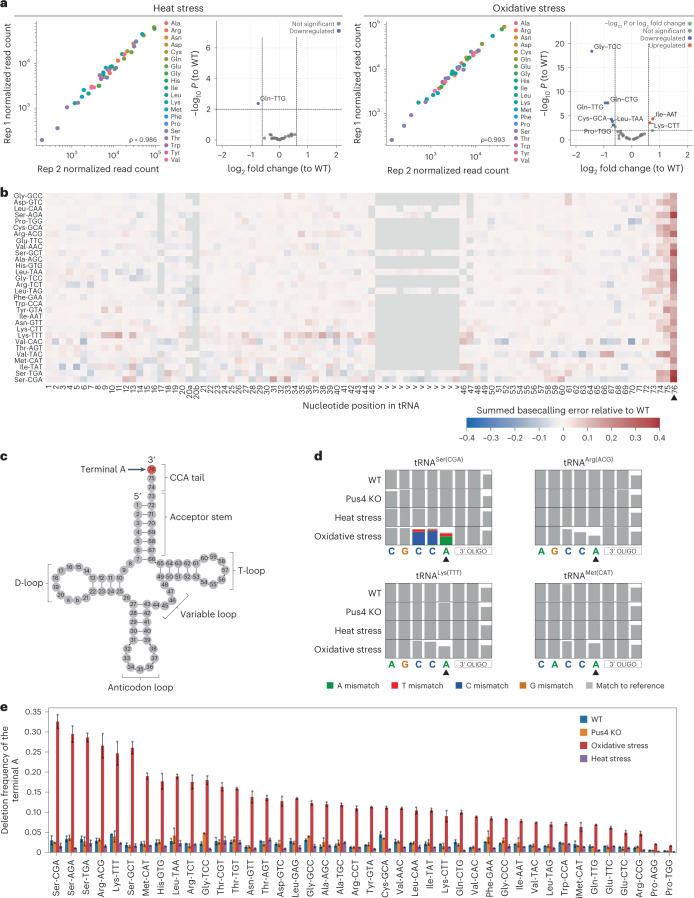
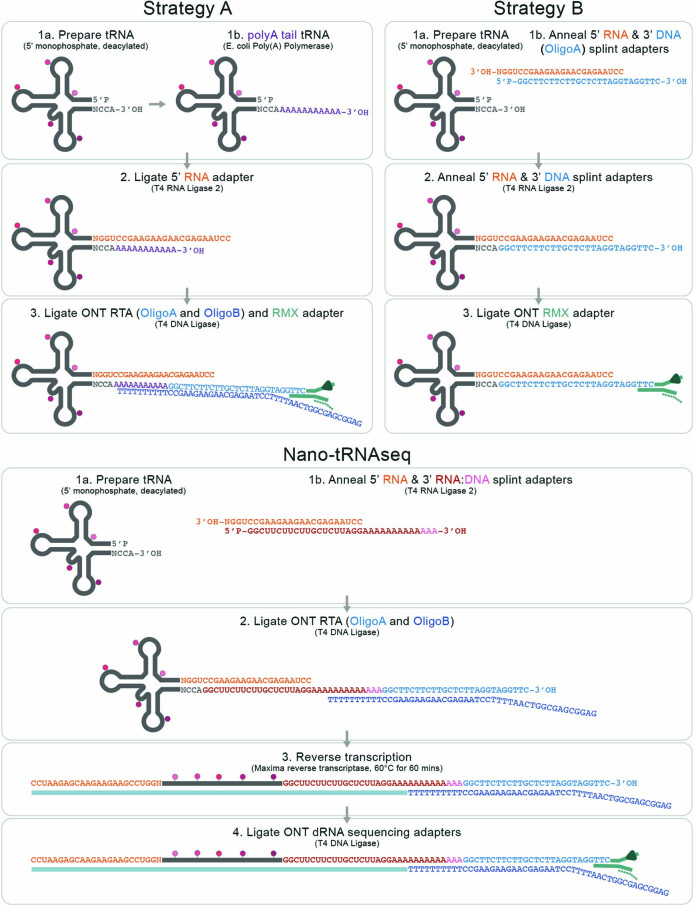
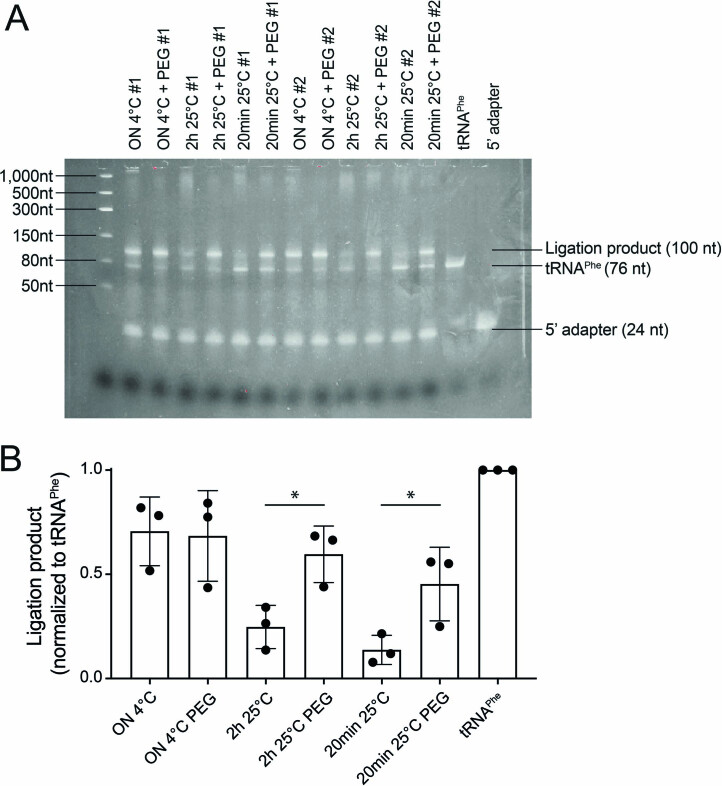
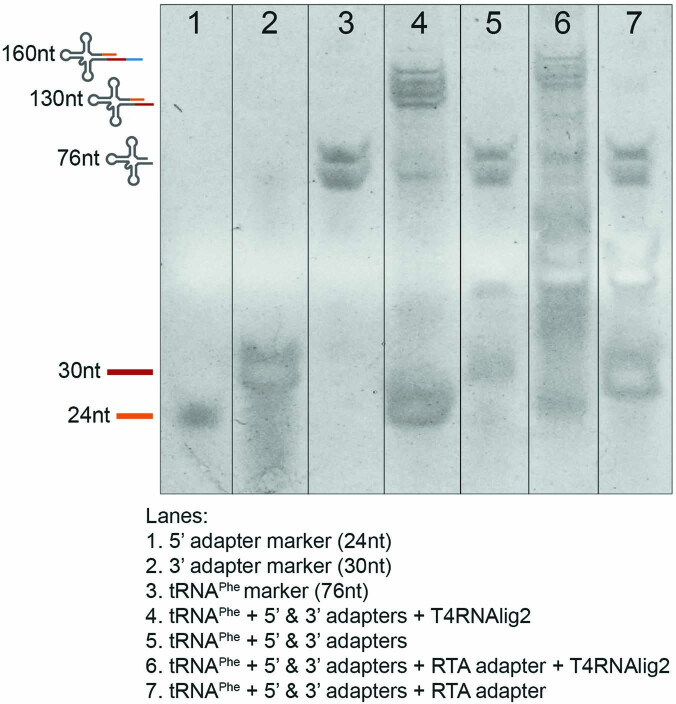
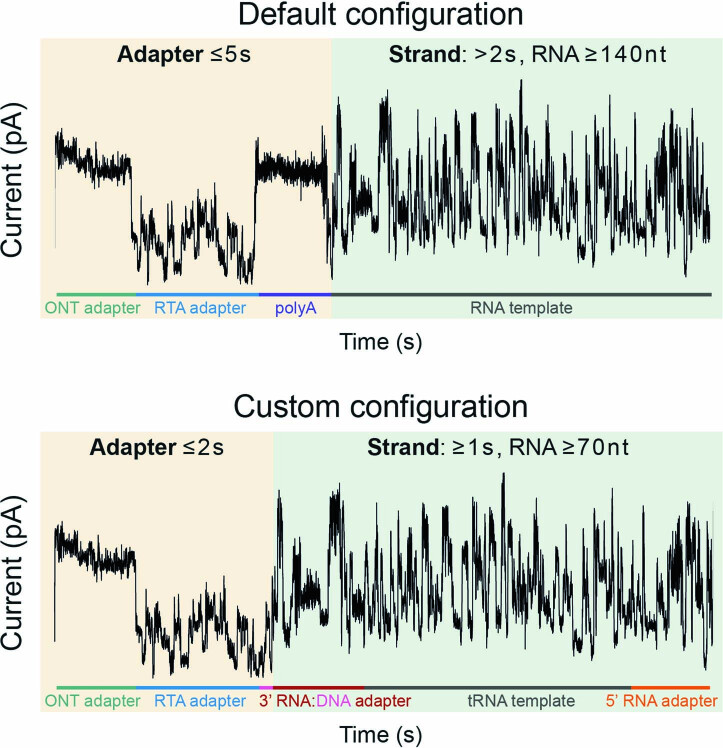
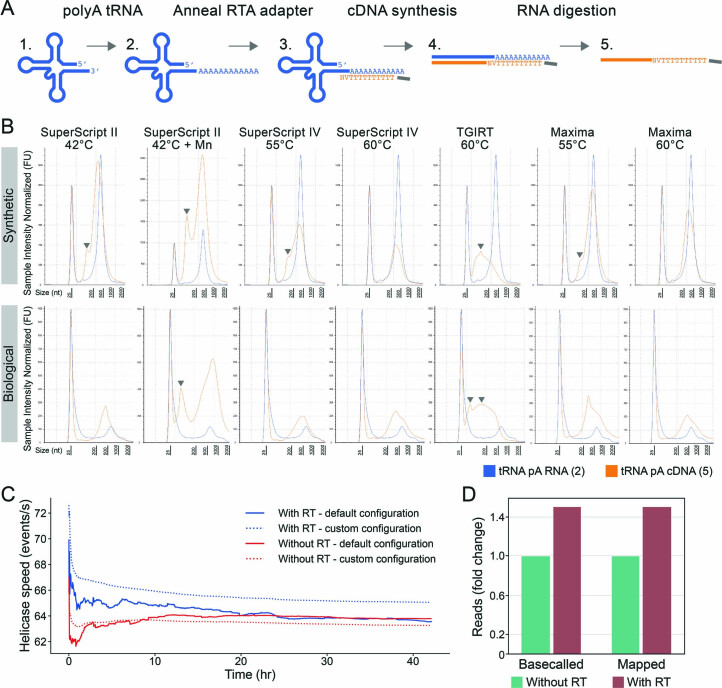
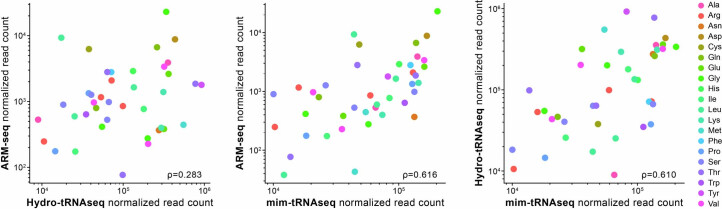
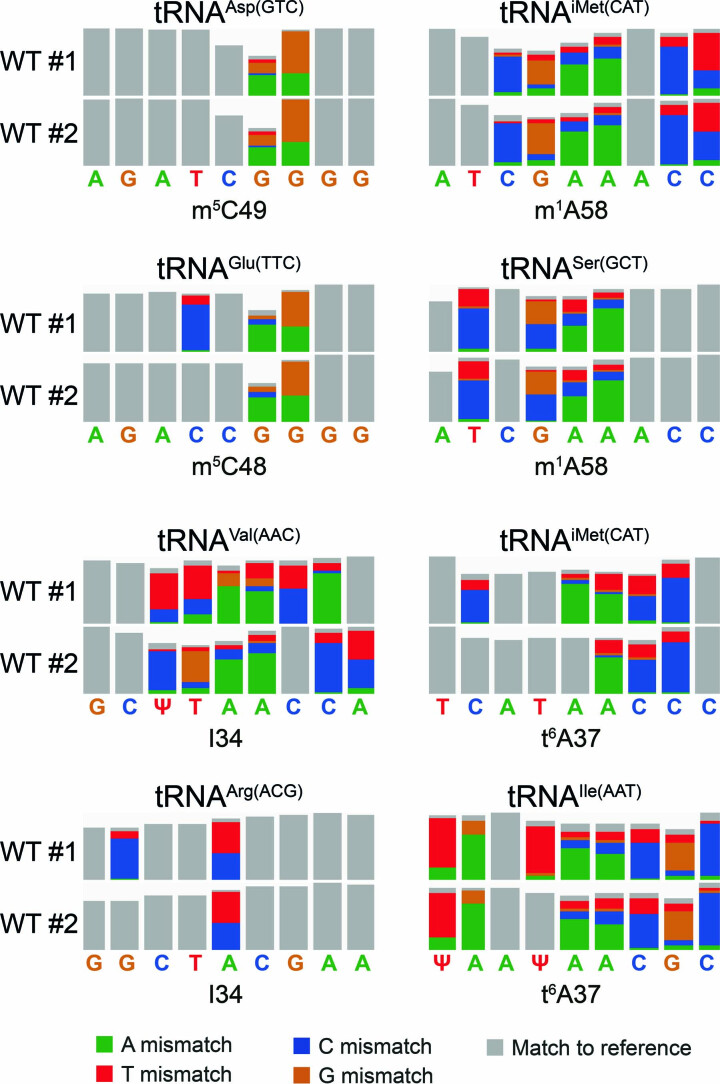
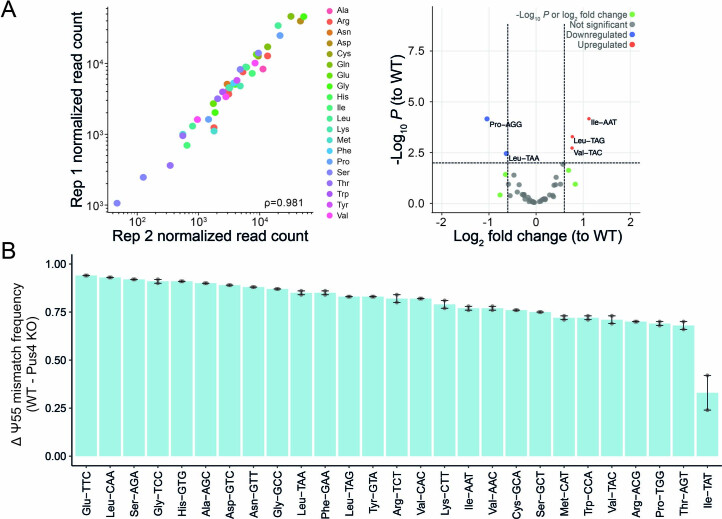
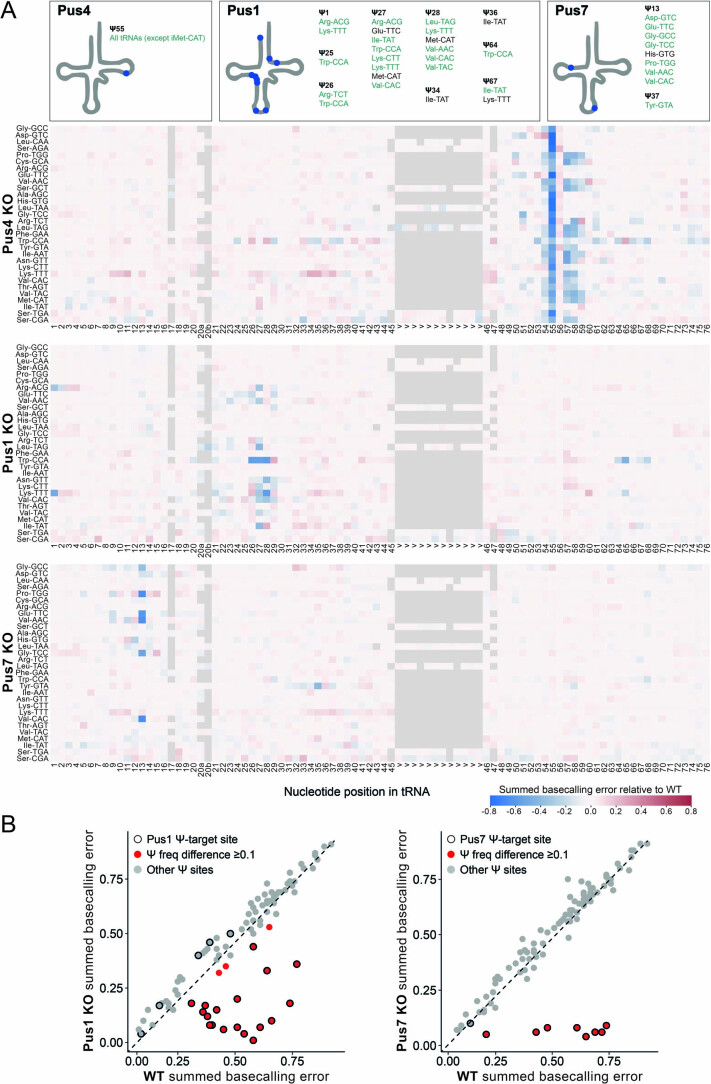
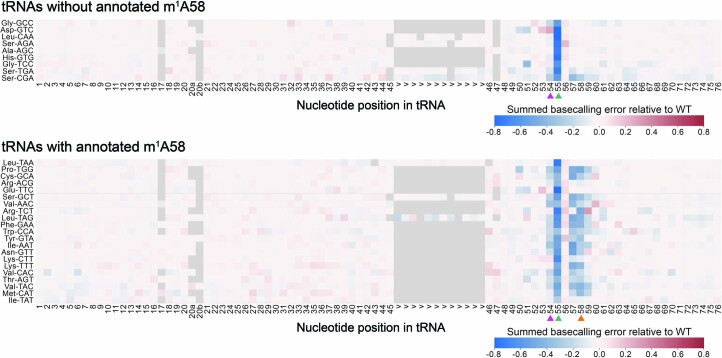
Transfer RNAs (tRNAs) play a central role in protein translation. Studying them has been difficult in part because a simple method to simultaneously quantify their abundance and chemical modifications is lacking. Here we introduce Nano-tRNAseq, a nanopore-based approach to sequence native tRNA populations that provides quantitative estimates of both tRNA abundances and modification dynamics in a single experiment. We show that default nanopore sequencing settings discard the vast majority of tRNA reads, leading to poor sequencing yields and biased representations of tRNA abundances based on their transcript length. Re-processing of raw nanopore current intensity signals leads to a 12-fold increase in the number of recovered tRNA reads and enables recapitulation of accurate tRNA abundances. We then apply Nano-tRNAseq to Saccharomyces cerevisiae tRNA populations, revealing crosstalks and interdependencies between different tRNA modification types within the same molecule and changes in tRNA populations in response to oxidative stress.
© 2023. The Author(s).
Conflict of interest statement
M.C.L., L.P. and E.M.N. have filed a patent on the Nano-tRNAseq library preparation method (application EP22382917). E.M.N. is a member of the Scientific Advisory Board of IMMAGINA Biotech. E.M.N. has received travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences, and M.C.L. has received an Oxford Nanopore Technologies travel bursary. The authors declare that the submitted work was otherwise carried out in the absence of any professional or financial relationships that could potentially be construed as a conflict of interest.
Figures
Comment in
-
Quantifying tRNA abundance by sequencing.Nat Genet. 2023 May;55(5):727. doi: 10.1038/s41588-023-01404-z. Nat Genet. 2023. PMID: 37173525 No abstract available.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
