

Gaps in Cystic Fibrosis Care Are Associated with Reduced Lung Function in the U.S. Cystic Fibrosis Foundation Patient Registry
- PMID: 37027571
- PMCID: PMC12039897
- DOI: 10.1513/AnnalsATS.202211-951OC
Gaps in Cystic Fibrosis Care Are Associated with Reduced Lung Function in the U.S. Cystic Fibrosis Foundation Patient Registry
Abstract
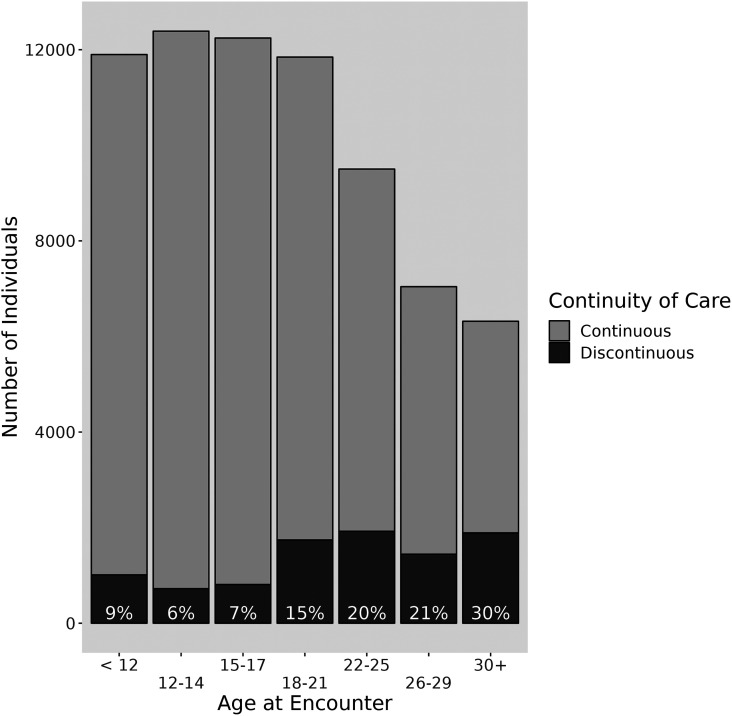
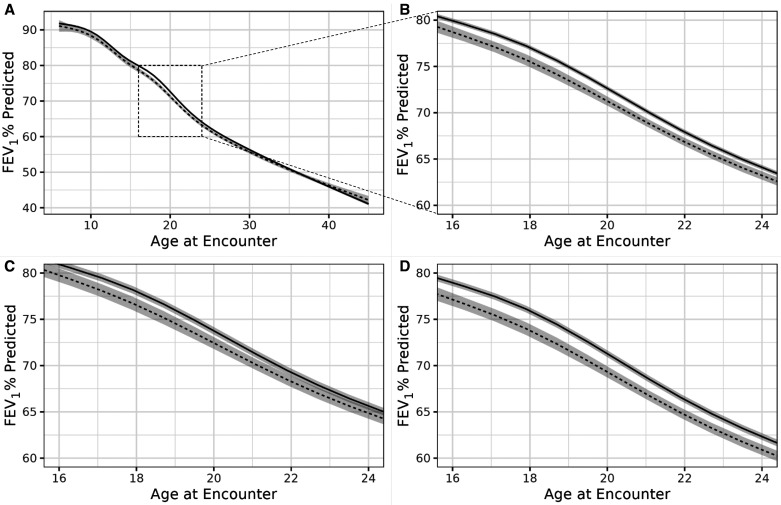
Rationale: Cystic fibrosis (CF) is a genetic disease leading to progressive lung function loss and early mortality. Many clinical and demographic variables are associated with lung function decline, but little is known about the effects of prolonged periods of missed care. Objectives: To determine if missed care in the Cystic Fibrosis Foundation Patient Registry (CFFPR) is associated with decreased lung function at follow-up visits. Methods: Deidentified CFFPR data for 2004-2016 were analyzed, with the exposure of interest being ⩾12-month gap in CFFPR data. We modeled percentage predicted forced expiratory volume in 1 second using longitudinal semiparametric modeling with natural cubic splines for age (knots at quantiles) and with subject-specific random effects, adjusted for sex and CFTR (cystic fibrosis transmembrane conductance regulator) genotype, race, and ethnicity and included time-varying covariates for gaps in care, insurance type, underweight body mass index, CF-related diabetes status, and chronic infections. Results: A total of 24,328 individuals with 1,082,899 encounters in the CFFPR met inclusion criteria. In the cohort, 8,413 (35%) individuals had at least a single ⩾12-month episode of discontinuity, whereas 15,915 (65%) had continuous care. Of the encounters preceded by a 12-month gap, 75.8% occurred in patients 18 years and older. Compared with those with continuous care, those with a discontinuous care episode had a lower follow-up percentage predicted forced expiratory volume in 1 second at the index visit (-0.81%; 95% confidence interval, -1.00, -0.61) after adjustment for other variables. The magnitude of this difference was much greater (-2.1%; 95% confidence interval, -1.5, -2.7) in young adult F508del homozygotes. Conclusions: There was a high rate of ⩾12-month gap in care, especially in adults, documented in the CFFPR. Discontinuous care identified in the CFFPR was strongly associated with decreased lung function, especially in adolescents and young adults homozygous for the F508del CFTR mutation. This may have implications for identifying and treating people with lengthy gaps in care and may have implications for CFF care recommendations.
Keywords: care fragmentation; cystic fibrosis; lung function decline.
Figures

Comment in
-
Falling through the Cracks-The Impact of Care Gaps on Lung Function Loss in Cystic Fibrosis.Ann Am Thorac Soc. 2023 Sep;20(9):1235-1236. doi: 10.1513/AnnalsATS.202305-459ED. Ann Am Thorac Soc. 2023. PMID: 37655958 Free PMC article. No abstract available.
Similar articles
-
Correctors (specific therapies for class II CFTR mutations) for cystic fibrosis.Cochrane Database Syst Rev. 2018 Aug 2;8(8):CD010966. doi: 10.1002/14651858.CD010966.pub2. Cochrane Database Syst Rev. 2018. Update in: Cochrane Database Syst Rev. 2020 Dec 17;12:CD010966. doi: 10.1002/14651858.CD010966.pub3. PMID: 30070364 Free PMC article. Updated. Review.
-
Impact of the expanded label for elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis with no F508del variant in the USA.Eur Respir J. 2024 Nov 14;64(5):2401146. doi: 10.1183/13993003.01146-2024. Print 2024 Nov. Eur Respir J. 2024. PMID: 39227072 Free PMC article.
-
Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data.J Cyst Fibros. 2018 Mar;17(2):218-227. doi: 10.1016/j.jcf.2017.11.019. Epub 2018 Jan 6. J Cyst Fibros. 2018. PMID: 29311001 Free PMC article.
-
Disease burden in people with cystic fibrosis heterozygous for F508del and a minimal function mutation.J Cyst Fibros. 2022 Jan;21(1):96-103. doi: 10.1016/j.jcf.2021.07.003. Epub 2021 Jul 19. J Cyst Fibros. 2022. PMID: 34289939 Free PMC article.
-
Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del).Cochrane Database Syst Rev. 2020 Dec 17;12(12):CD010966. doi: 10.1002/14651858.CD010966.pub3. Cochrane Database Syst Rev. 2020. Update in: Cochrane Database Syst Rev. 2023 Nov 20;11:CD010966. doi: 10.1002/14651858.CD010966.pub4. PMID: 33331662 Free PMC article. Updated.
Cited by
-
Observational Studies.Respir Care. 2023 Nov;68(11):1585-1597. doi: 10.4187/respcare.11170. Epub 2023 Jun 20. Respir Care. 2023. PMID: 37339891 Free PMC article.
-
Predictors of frequency of CF care in the US Cystic Fibrosis Foundation Patient Registry.PLoS One. 2024 Dec 3;19(12):e0313510. doi: 10.1371/journal.pone.0313510. eCollection 2024. PLoS One. 2024. PMID: 39625878 Free PMC article.
-
Health Care Transitions Among Adolescents and Young Adults With Cancer.J Clin Oncol. 2024 Feb 20;42(6):743-754. doi: 10.1200/JCO.23.01504. Epub 2024 Jan 9. J Clin Oncol. 2024. PMID: 38194608 Free PMC article. Review.
-
Falling through the Cracks-The Impact of Care Gaps on Lung Function Loss in Cystic Fibrosis.Ann Am Thorac Soc. 2023 Sep;20(9):1235-1236. doi: 10.1513/AnnalsATS.202305-459ED. Ann Am Thorac Soc. 2023. PMID: 37655958 Free PMC article. No abstract available.
-
Cystic fibrosis year in review 2023.Pediatr Pulmonol. 2024 Dec;59(12):3106-3116. doi: 10.1002/ppul.27190. Epub 2024 Jul 26. Pediatr Pulmonol. 2024. PMID: 39056532 Free PMC article. Review.
References
-
- Kerem E, Reisman J, Corey M, Canny GJ, Levison H. Prediction of mortality in patients with cystic fibrosis. N Engl J Med . 1992;326:1187–1191. - PubMed
-
- Knapp EA, Fink AK, Goss CH, Sewall A, Ostrenga J, Dowd C, et al. The Cystic Fibrosis Foundation Patient Registry. Design and methods of a national observational disease registry. Ann Am Thorac Soc . 2016;13:1173–1179. - PubMed
-
- Cystic Fibrosis Foundation. 2021. https://www.cff.org/managing-cf/cf-care-center-visits#:~:text=CF%20Found...
-
- Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D. Cystic fibrosis adult care: consensus conference report. Chest . 2004;125:1S–39S. - PubMed
-
- Schaedel C, de Monestrol I, Hjelte L, Johannesson M, Kornfält R, Lindblad A, et al. Predictors of deterioration of lung function in cystic fibrosis. Pediatr Pulmonol . 2002;33:483–491. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
