

Recent insights into lysosomal acid lipase deficiency
- PMID: 37028992
- PMCID: PMC7614602
- DOI: 10.1016/j.molmed.2023.03.001
Recent insights into lysosomal acid lipase deficiency
Abstract
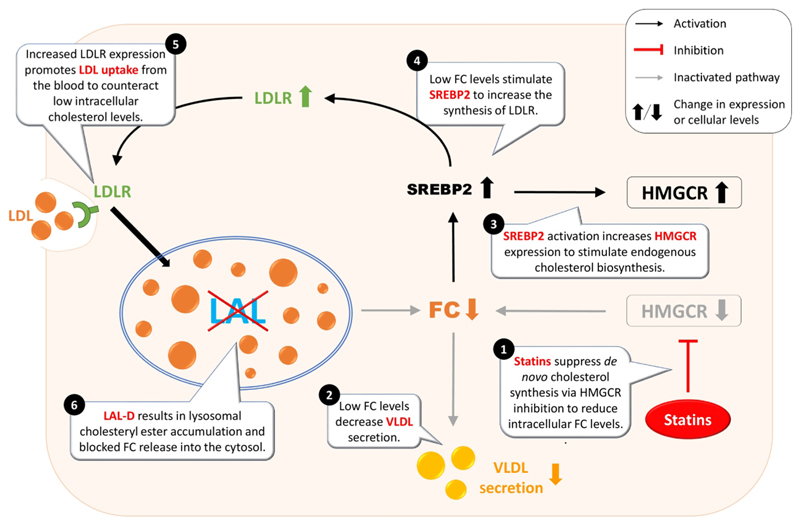
Lysosomal acid lipase (LAL) is the sole enzyme known to degrade neutral lipids in the lysosome. Mutations in the LAL-encoding LIPA gene lead to rare lysosomal lipid storage disorders with complete or partial absence of LAL activity. This review discusses the consequences of defective LAL-mediated lipid hydrolysis on cellular lipid homeostasis, epidemiology, and clinical presentation. Early detection of LAL deficiency (LAL-D) is essential for disease management and survival. LAL-D must be considered in patients with dyslipidemia and elevated aminotransferase concentrations of unknown etiology. Enzyme replacement therapy, sometimes in combination with hematopoietic stem cell transplantation (HSCT), is currently the only therapy for LAL-D. New technologies based on mRNA and viral vector gene transfer are recent efforts to provide other effective therapeutic strategies.
Keywords: LAL-D; LIPA; Wolman disease; acid lipolysis; cholesteryl ester storage disease; lysosomal lipid storage disorder.
Copyright © 2023 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests No interests are declared.
Figures
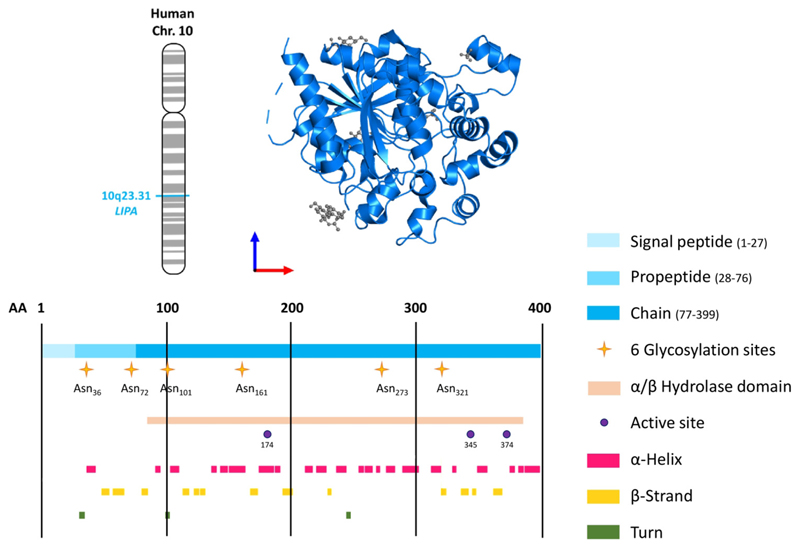
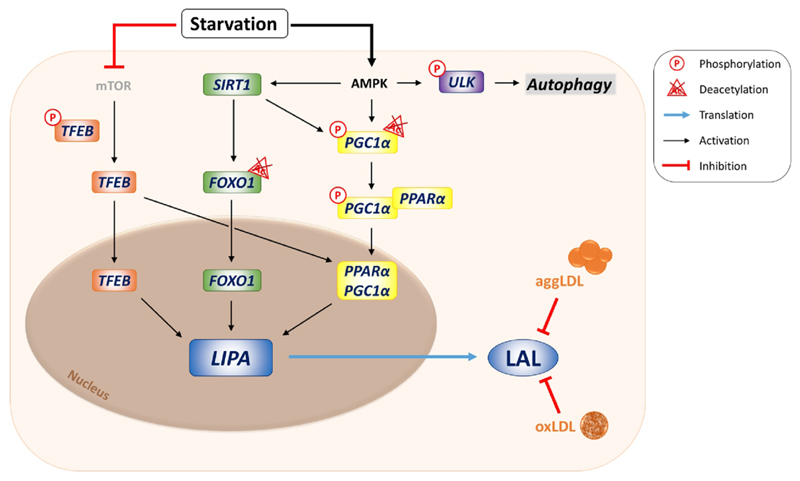
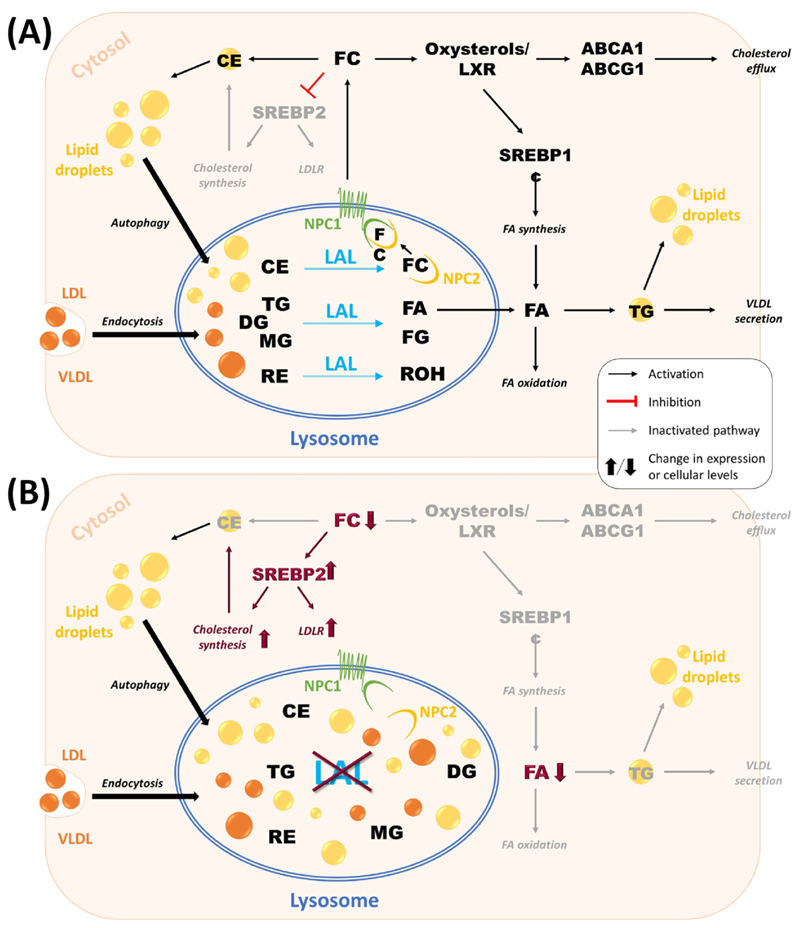
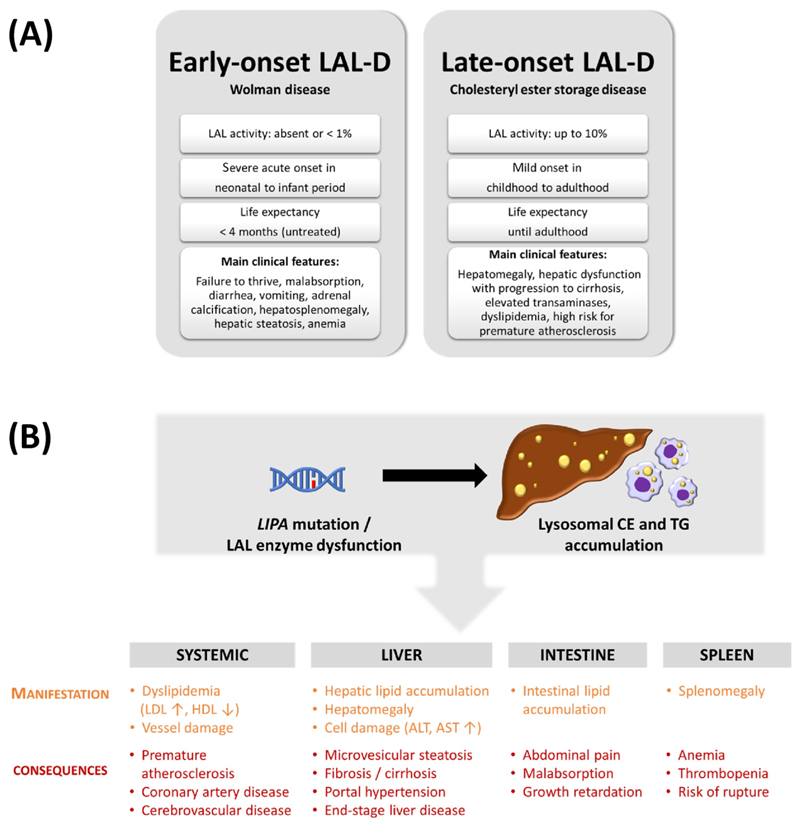
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
