

Scientific rationale for the use of α2A-adrenoceptor agonists in treating neuroinflammatory cognitive disorders
- PMID: 37029295
- PMCID: PMC10080530
- DOI: 10.1038/s41380-023-02057-4
Scientific rationale for the use of α2A-adrenoceptor agonists in treating neuroinflammatory cognitive disorders
Abstract
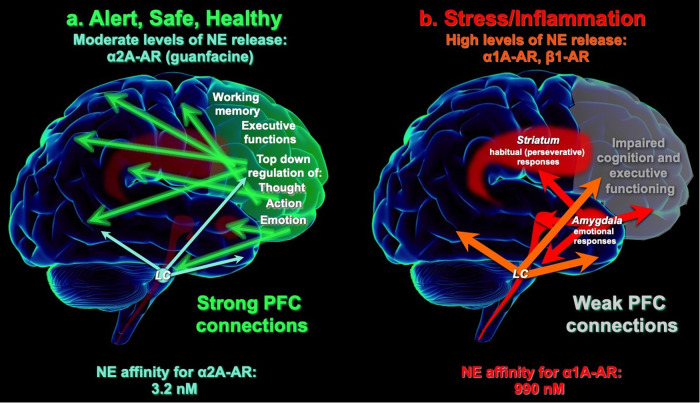
Neuroinflammatory disorders preferentially impair the higher cognitive and executive functions of the prefrontal cortex (PFC). This includes such challenging disorders as delirium, perioperative neurocognitive disorder, and the sustained cognitive deficits from "long-COVID" or traumatic brain injury. There are no FDA-approved treatments for these symptoms; thus, understanding their etiology is important for generating therapeutic strategies. The current review describes the molecular rationale for why PFC circuits are especially vulnerable to inflammation, and how α2A-adrenoceptor (α2A-AR) actions throughout the nervous and immune systems can benefit the circuits in PFC needed for higher cognition. The layer III circuits in the dorsolateral PFC (dlPFC) that generate and sustain the mental representations needed for higher cognition have unusual neurotransmission and neuromodulation. They are wholly dependent on NMDAR neurotransmission, with little AMPAR contribution, and thus are especially vulnerable to kynurenic acid inflammatory signaling which blocks NMDAR. Layer III dlPFC spines also have unusual neuromodulation, with cAMP magnification of calcium signaling in spines, which opens nearby potassium channels to rapidly weaken connectivity and reduce neuronal firing. This process must be tightly regulated, e.g. by mGluR3 or α2A-AR on spines, to prevent loss of firing. However, the production of GCPII inflammatory signaling reduces mGluR3 actions and markedly diminishes dlPFC network firing. Both basic and clinical studies show that α2A-AR agonists such as guanfacine can restore dlPFC network firing and cognitive function, through direct actions in the dlPFC, but also by reducing the activity of stress-related circuits, e.g. in the locus coeruleus and amygdala, and by having anti-inflammatory actions in the immune system. This information is particularly timely, as guanfacine is currently the focus of large clinical trials for the treatment of delirium, and in open label studies for the treatment of cognitive deficits from long-COVID.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
Stress and Inflammation Target Dorsolateral Prefrontal Cortex Function: Neural Mechanisms Underlying Weakened Cognitive Control.Biol Psychiatry. 2025 Feb 15;97(4):359-371. doi: 10.1016/j.biopsych.2024.06.016. Epub 2024 Jun 27. Biol Psychiatry. 2025. PMID: 38944141 Review.
-
Guanfacine's mechanism of action in treating prefrontal cortical disorders: Successful translation across species.Neurobiol Learn Mem. 2020 Dec;176:107327. doi: 10.1016/j.nlm.2020.107327. Epub 2020 Oct 17. Neurobiol Learn Mem. 2020. PMID: 33075480 Free PMC article. Review.
-
Scientific Rationale for the Treatment of Cognitive Deficits from Long COVID.Neurol Int. 2023 May 31;15(2):725-742. doi: 10.3390/neurolint15020045. Neurol Int. 2023. PMID: 37368329 Free PMC article. Review.
-
Inhibition of glutamate-carboxypeptidase-II in dorsolateral prefrontal cortex: potential therapeutic target for neuroinflammatory cognitive disorders.Mol Psychiatry. 2022 Oct;27(10):4252-4263. doi: 10.1038/s41380-022-01656-x. Epub 2022 Jun 22. Mol Psychiatry. 2022. PMID: 35732693 Free PMC article.
-
Unique Molecular Regulation of Higher-Order Prefrontal Cortical Circuits: Insights into the Neurobiology of Schizophrenia.ACS Chem Neurosci. 2018 Sep 19;9(9):2127-2145. doi: 10.1021/acschemneuro.7b00505. Epub 2018 Mar 1. ACS Chem Neurosci. 2018. PMID: 29470055 Free PMC article. Review.
Cited by
-
Dexmedetomidine potently and reversibly regulates stress-mediated behaviors.Front Pharmacol. 2025 Jul 2;16:1589075. doi: 10.3389/fphar.2025.1589075. eCollection 2025. Front Pharmacol. 2025. PMID: 40672361 Free PMC article.
-
Successful treatment with guanfacine in a long-COVID case manifesting marked cognitive impairment.Neuropsychopharmacol Rep. 2024 Sep;44(3):585-590. doi: 10.1002/npr2.12466. Epub 2024 Jun 27. Neuropsychopharmacol Rep. 2024. PMID: 38934345 Free PMC article.
-
Activation of α 2A and α 2B -adrenergic receptors inhibits tactile stimulation-evoked parallel fiber-Purkinje cell synaptic transmission in mouse cerebellar cortex.Neuroreport. 2024 Feb 7;35(2):115-122. doi: 10.1097/WNR.0000000000001983. Epub 2023 Dec 12. Neuroreport. 2024. PMID: 38109417 Free PMC article.
-
The Biology and Biochemistry of Kynurenic Acid, a Potential Nutraceutical with Multiple Biological Effects.Int J Mol Sci. 2024 Aug 21;25(16):9082. doi: 10.3390/ijms25169082. Int J Mol Sci. 2024. PMID: 39201768 Free PMC article. Review.
-
Stress and Inflammation Target Dorsolateral Prefrontal Cortex Function: Neural Mechanisms Underlying Weakened Cognitive Control.Biol Psychiatry. 2025 Feb 15;97(4):359-371. doi: 10.1016/j.biopsych.2024.06.016. Epub 2024 Jun 27. Biol Psychiatry. 2025. PMID: 38944141 Review.
References
-
- McDonald B, Flashman L, Saykin AJ. Executive dysfunction following traumatic brain injury: neural substrates and treatment strategies. NeuroRehabilitation. 2002;17:33–44.. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
